The relationship between sponsors and contract organizations has evolved far beyond simple transactional exchanges. Digital infrastructure has become the cornerstone of trust, transparency, and operational excellence.
The trust equation is fundamentally changing due to the way our supply chains are being challenged.. Traditional quality agreements often functioned as static documents—comprehensive but disconnected from day-to-day operations. Today’s most successful partnerships are built on dynamic, digitally-enabled frameworks that provide real-time visibility into performance, compliance, and risk management.
Regulatory agencies are increasingly scrutinizing the effectiveness of sponsor oversight programs. The FDA’s emphasis on data integrity, combined with EMA’s evolving computerized systems requirements, means that sponsors can no longer rely on periodic audits and static documentation to demonstrate control over their outsourced activities.
Quality Agreements as Digital Trust Frameworks
The modern quality agreement must evolve from a compliance document to a digital trust framework. This transformation requires reimagining three fundamental components:
Dynamic Risk Assessment Integration
Traditional quality agreements categorize suppliers into static risk tiers (for example Category 1, 2, 2.5, or 3 based on material/service risk). Digital frameworks enable continuous risk profiling that adapts based on real-time performance data.
Integrate supplier performance metrics directly into your quality management system. When a Category 2 supplier’s on-time delivery drops below threshold or quality metrics deteriorate, the system should automatically trigger enhanced monitoring protocols without waiting for the next periodic review.
Automated Change Control Workflows
One of the most contentious areas in sponsor-CxO relationships involves change notifications and approvals. Digital infrastructure can transform this friction point into a competitive advantage.
The SMART approach to change control:
Standardized digital templates for change notifications
Machine-readable impact assessments
Automated routing based on change significance
Real-time status tracking for all stakeholders
Traceable decision logs with electronic signatures
Quality agreement language to include: “All change notifications shall be submitted through the designated digital platform within [X] business days of identification, with automated acknowledgment and preliminary impact assessment provided within [Y] hours.”
Transparent Performance Dashboards
The most innovative CxOs are moving beyond quarterly business reviews to continuous performance visibility. Quality agreements should build upon real-time access to key performance indicators (KPIs) that matter most to patient safety and product quality.
Examples of Essential KPIs for digital dashboards:
Batch disposition times and approval rates
Deviation investigation cycle times
CAPA effectiveness metrics
Environmental monitoring excursions and response times
Supplier change notification compliance rates
Communication Architecture for Transparency
Effective communication in pharmaceutical partnerships requires architectural thinking, not just protocol definition. The most successful CxO-sponsor relationships are built on what I call the “Three-Layer Communication Stack” which builds a rhythm of communication:
Layer 1: Operational Communication (Real-Time)
Purpose: Day-to-day coordination and issue resolution
Tools: Integrated messaging within quality management systems, automated alerts, mobile notifications
Quality agreement requirement: “Operational communications shall be conducted through validated, audit-trailed platforms with 24/7 availability and guaranteed delivery confirmation.”
Every quality agreement should include a subsidiary Communication Plan that addresses:
Stakeholder Matrix: Who needs what information, when, and in what format
Escalation Protocols: Clear triggers for moving issues up the communication stack
Performance Metrics: How communication effectiveness will be measured and improved
Technology Requirements: Specified platforms, security requirements, and access controls
Contingency Procedures: Alternative communication methods for system failures or emergencies
Include communication effectiveness as a measurable element in your supplier scorecards. Track metrics like response time to quality notifications, accuracy of status reporting, and proactive problem identification.
Data Governance as a Competitive Differentiator
Data integrity is more than just ensuring ALCOA+—it’s about creating a competitive moat through superior data governance. The organizations that master data sharing, analysis, and decision-making will dominate the next decade of pharmaceutical manufacturing and development.
The Modern Data Governance Framework
Data Architecture Definition
Your quality agreement must specify not just what data will be shared, but how it will be structured, validated, and integrated:
Master data management: Consistent product codes, batch numbering, and material identifiers across all systems
Data quality standards: Validation rules, completeness requirements, and accuracy thresholds
Integration protocols: APIs, data formats, and synchronization frequencies
With increasing regulatory focus on cybersecurity, your data governance plan must address:
Role-based access controls: Granular permissions based on job function and business need
Data classification: Confidentiality levels and handling requirements
Audit logging: Comprehensive tracking of data access, modification, and sharing
Analytics and Intelligence
The real competitive advantage comes from turning shared data into actionable insights:
Predictive analytics: Early warning systems for quality trends and supply chain disruptions
Benchmark reporting: Anonymous industry comparisons to identify improvement opportunities
Root cause analysis: Automated correlation of events across multiple systems and suppliers
The Data Governance Subsidiary Agreement
Consider creating a separate Data Governance Agreement that complements your quality agreement with specific sections covering data sharing objectives, technical architecture, governance oversight, and compliance requirements.
Veeva Summit
Next week I’ll be discussing this topic at the Veeva Summit, where I will bring some organizational learnings on to embrace digital infrastructure as a trust-building mechanism will forge stronger partnerships, achieve superior quality outcomes, and ultimately deliver better patient experiences.
If you are like me, you face a fundamental choice on a daily (or hourly basis): we can either develop distributed decision-making capability throughout our organizations, or we can create bottlenecks that compromise our ability to respond effectively to quality events, regulatory changes, and operational challenges. The reactive control mindset—where senior quality leaders feel compelled to personally approve every decision—creates dangerous delays in an industry where timing can directly impact patient safety.
It makes sense, we are an experience based profession, so decisions tend to need by more experienced people. But that can really lead to an over tendency to make decisions. Next time you are being asked to make a decision as these four questions.
1. Who is Closest to the Action?
Proximity is a form of expertise. The quality team member completing batch record reviews has direct insight into manufacturing anomalies that executive summaries cannot capture. The QC analyst performing environmental monitoring understands contamination patterns that dashboards obscure. The validation specialist working on equipment qualification sees risk factors that organizational charts miss.
Consider routine decisions about cleanroom environmental monitoring deviations. The microbiologist analyzing the data understands the contamination context, seasonal patterns, and process-specific risk factors better than any senior leader reviewing summary reports. When properly trained and given clear escalation criteria, they can make faster, more scientifically grounded decisions about investigation scope and corrective actions.
This connects directly to ICH Q9(R1)’s principle of formality in quality risk management. The level of delegation should be commensurate with the risk level, but routine decisions with established precedent and clear acceptance criteria represent prime candidates for systematic delegation.
3. Leveraging Specialized Expertise
In pharmaceutical quality, technical depth often trumps hierarchical position in decision quality. The microbiologist analyzing contamination events may have specialized knowledge that outweighs organizational seniority. The specialist tracking FDA guidance may see compliance implications that escape broader quality leadership attention.
Consider biologics manufacturing decisions where process characterization data must inform manufacturing parameters. The bioprocess engineer analyzing cell culture performance data possesses specialized insight that generic quality management cannot match. When decision authority is properly structured, these technical experts can make more informed decisions about process adjustments within validated ranges.
4. Eliminating Decision Bottlenecks
Quality systems are particularly vulnerable to momentum-stalling bottlenecks. CAPA timelines extend, investigations languish, and validation activities await approvals because decision authority remains unclear. In our regulated environment, the risk isn’t just a suboptimal decision—it’s often no decision at all, which can create far greater compliance and patient safety risks.
Contamination control strategies, environmental monitoring programs, and cleaning validation protocols all suffer when every decision must flow through senior quality leadership. Strategic delegation creates clear authority for qualified team members to act within defined parameters while maintaining appropriate oversight.
Building Decision Architecture in Quality Systems
Effective delegation in pharmaceutical quality requires systematic implementation:
Phase 1: Decision Mapping and Risk Assessment
Using quality risk management principles, catalog your current decision types:
High-risk, infrequent decisions: Major CAPA approvals, manufacturing process changes, regulatory submission decisions (retain centralized authority)
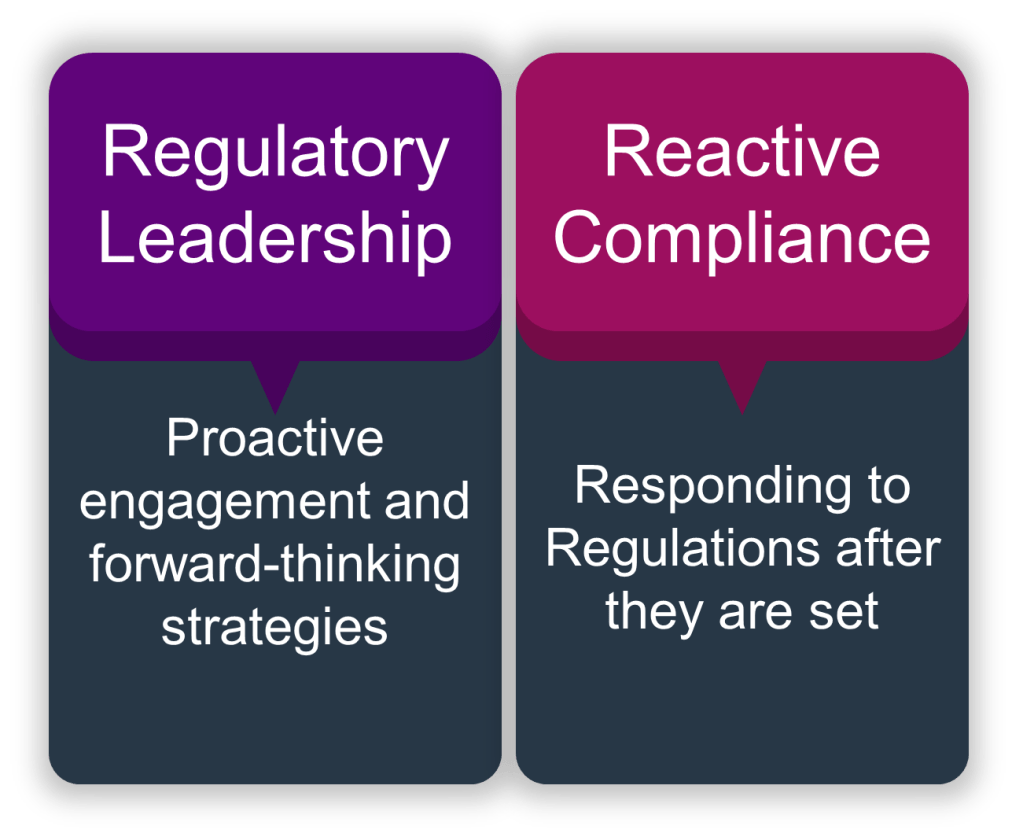
In pharmaceutical quality, we face a fundamental choice that defines our trajectory: we can either help set the direction of our regulatory landscape, or we can struggle to keep up with changes imposed upon us. As quality leaders, this choice isn’t just about compliance—it’s about positioning our organizations to drive meaningful change while delivering better patient outcomes.
The reactive compliance mindset has dominated our industry for too long, where companies view regulators as adversaries and quality as a cost center. This approach treats regulatory guidance as something that happens to us rather than something we actively shape. Companies operating in this mode find themselves perpetually behind the curve, scrambling to interpret new requirements, implement last-minute changes, and justify their approaches to skeptical regulators.
But there’s another way—one where quality professionals actively engage with the regulatory ecosystem to influence the development of standards before they become mandates.
The Strategic Value of Industry Group Engagement
Organizations like BioPhorum, NIIMBL, ISPE, and PDA represent far more than networking opportunities—they are the laboratories where tomorrow’s regulatory expectations are forged today. These groups don’t just discuss new regulations; they actively participate in defining what excellence looks like through standard-setting initiatives, white papers, and direct dialogue with regulatory authorities.
BioPhorum, with its collaborative network of 160+ manufacturers and suppliers deploying over 7,500 subject matter experts, demonstrates the power of collective engagement. Their success stories speak to tangible outcomes: harmonized approaches to routine environmental monitoring that save weeks on setup time, product yield improvements of up to 44%, and flexible manufacturing lines that reduce costs while maintaining regulatory compliance. Most significantly, their quality phorum launched in 2024 provides a dedicated space for quality professionals to collaborate on shared industry challenges.
NIIMBL exemplifies the strategic integration of industry voices with federal priorities, bringing together pharmaceutical manufacturers with academic institutions and government agencies to advance biopharmaceutical manufacturing standards. Their public-private partnership model demonstrates how industry engagement can shape policy while advancing technical capabilities that benefit all stakeholders.
ISPE and PDA provide complementary platforms where technical expertise translates into regulatory influence. Through their guidance documents, technical reports, and direct responses to regulatory initiatives, these organizations ensure that industry perspectives inform regulatory development. Their members don’t just consume regulatory intelligence—they help create it.
The Big Company Advantage—And Why Smaller Companies Must Close This Gap
Large pharmaceutical companies understand this dynamic intuitively. They maintain dedicated teams whose sole purpose is to engage with these industry groups, contribute to standard-setting activities, and maintain ongoing relationships with regulatory authorities. They recognize that regulatory intelligence isn’t just about monitoring changes—it’s about influencing the trajectory of those changes before they become requirements.
The asymmetry is stark: while multinational corporations deploy key leaders to these forums, smaller innovative companies often view such engagement as a luxury they cannot afford. This creates a dangerous gap where the voices shaping regulatory policy come predominantly from established players, potentially disadvantaging the very companies driving the most innovative therapeutic approaches.
But here’s the critical insight from my experience working with quality systems: smaller companies cannot afford NOT to be at these tables. When you’re operating with limited resources, you need every advantage in predicting regulatory direction, understanding emerging expectations, and building the credibility that comes from being recognized as a thoughtful contributor to industry discourse.
Consider the TESTED framework I’ve previously discussed—structured hypothesis formation requires deep understanding of regulatory thinking that only comes from being embedded in these conversations. When BioPhorum members collaborate on cleaning validation approaches or manufacturing flexibility standards, they’re not just sharing best practices—they’re establishing the scientific foundation for future regulatory expectations. When the ISPE comes out with a new good practice guide they are doing the same. The list goes on.
Making the Business Case: Job Descriptions and Performance Evaluation
Good regulatory intelligence practices requires systematically building this engagement into our organizational DNA. This means making industry participation an explicit component of senior quality roles and measuring our leaders’ contributions to the broader regulatory dialogue.
For quality directors and above, job descriptions should explicitly include:
Active participation in relevant industry working groups and technical committees
Contribution to industry white papers, guidance documents, and technical reports
Maintenance of productive relationships with regulatory authorities through formal and informal channels
Intelligence gathering and strategic assessment of emerging regulatory trends
Internal education and capability building based on industry insights
Performance evaluations must reflect these priorities:
Measure contributions to industry publications and standard-setting activities
Assess the quality and strategic value of regulatory intelligence gathered through industry networks
Evaluate success in anticipating and preparing for regulatory changes before they become requirements
Track the organization’s reputation within industry forums as a thoughtful contributor
This isn’t about checking boxes or accumulating conference attendance credits. It’s about recognizing that in our interconnected regulatory environment, isolation equals irrelevance. The companies that will thrive in tomorrow’s regulatory landscape are those whose leaders are actively shaping that landscape today.
Development plans for individuals should have clear milestones based on these requirements, so as individuals work their way up in an organization they are building good behaviors.
The Competitive Advantage of Regulatory Leadership
When we engage strategically with industry groups, we gain access to three critical advantages that reactive companies lack. First, predictive intelligence—understanding not just what regulations say today, but where regulatory thinking is headed. Second, credibility capital—the trust that comes from being recognized as a thoughtful contributor rather than a passive recipient of regulatory requirements. Third, collaborative problem-solving—access to the collective expertise needed to address complex quality challenges that no single organization can solve alone.
The pharmaceutical industry is moving toward more sophisticated quality metrics, risk-based approaches, and integrated lifecycle management. Companies that help develop these approaches will implement them more effectively than those who wait for guidance to arrive as mandates.c
As I’ve explored in previous discussions of hypothesis-driven quality systems, the future belongs to organizations that can move beyond compliance toward genuine quality leadership. This requires not just technical excellence, but strategic engagement with the regulatory ecosystem that shapes our industry’s direction.
The choice is ours: we can continue struggling to keep up with changes imposed upon us, or we can help set the direction through strategic engagement with the organizations and forums that define excellence in our field. For senior quality leaders, this isn’t just a career opportunity—it’s a strategic imperative that directly impacts our organizations’ ability to deliver innovative therapies to patients who need them.
The bandwidth required for this engagement isn’t overhead—it’s investment in the intelligence and relationships that make everything else we do more effective. In a world where regulatory agility determines competitive advantage, being at the table where standards are set isn’t optional—it’s essential.
In my previous post, “The Effectiveness Paradox: Why ‘Nothing Bad Happened’ Doesn’t Prove Your Quality System Works”, I challenged a core assumption underpinning how the pharmaceutical industry evaluates its quality systems. We have long mistaken the absence of negative events—no deviations, no recalls, no adverse findings—for evidence of effectiveness. As I argued, this is not proof of success, but rather a logical fallacy: conflating absence of evidence with evidence of absence, and building unfalsifiable systems that teach us little about what truly works.
But recognizing the limits of “nothing bad happened” as a quality metric is only the starting point. The real challenge is figuring out what comes next: How do we move from defensive, unfalsifiable quality posturing toward a framework where our systems can be genuinely and rigorously tested? How do we design quality management approaches that not only anticipate success but are robust enough to survive—and teach us from—failure?
The answer begins with transforming the way we frame and test our assumptions about quality performance. Enter structured hypothesis formation: a disciplined, scientific approach that takes us from passive observation to active, falsifiable prediction. This methodology doesn’t just close the door on the effectiveness paradox—it opens a new frontier for quality decision-making grounded in scientific rigor, predictive learning, and continuous improvement.
The Science of Structured Hypothesis Formation
Structured hypothesis formation differs fundamentally from traditional quality planning by emphasizing falsifiability and predictive capability over compliance demonstration. Where traditional approaches ask “How can we prove our system works?” structured hypothesis formation asks “What specific predictions can our quality system make, and how can these predictions be tested?”
The core principles of structured hypothesis formation in quality systems include:
Explicit Prediction Generation: Quality hypotheses must make specific, measurable predictions about system behavior under defined conditions. Rather than generic statements like “our cleaning process prevents cross-contamination,” effective hypotheses specify conditions: “our cleaning procedure will reduce protein contamination below 10 ppm within 95% confidence when contact time exceeds 15 minutes at temperatures above 65°C.”
Testable Mechanisms: Hypotheses should articulate the underlying mechanisms that drive quality outcomes. This moves beyond correlation toward causation, enabling genuine process understanding rather than statistical association.
Failure Mode Specification: Effective quality hypotheses explicitly predict how and when systems will fail, creating opportunities for proactive detection and mitigation rather than reactive response.
Uncertainty Quantification: Rather than treating uncertainty as weakness, structured hypothesis formation treats uncertainty quantification as essential for making informed quality decisions under realistic conditions.
Framework for Implementation: The TESTED Approach
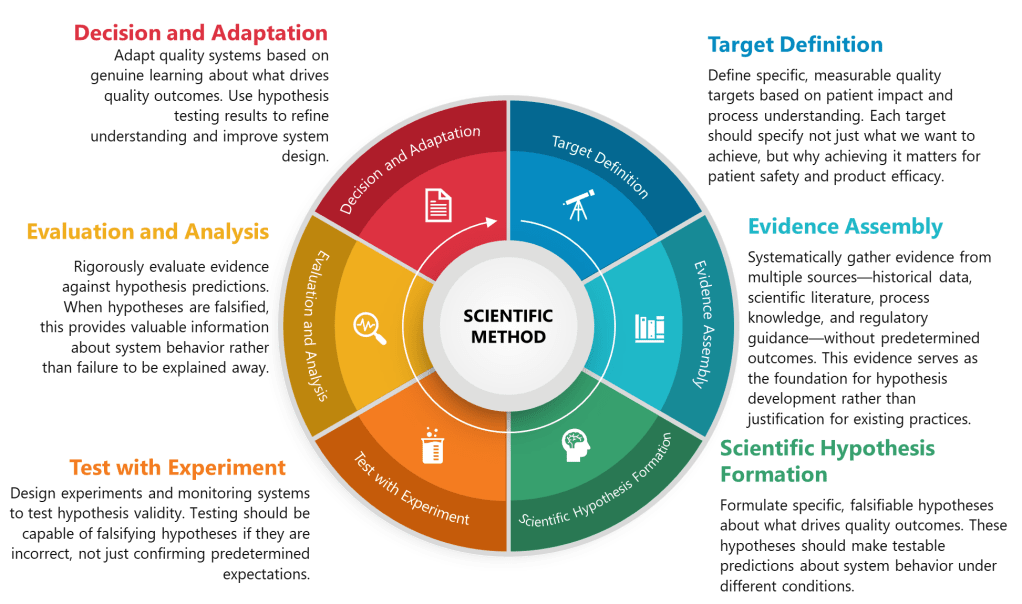
The practical implementation of structured hypothesis formation in pharmaceutical quality systems can be systematized through what I call the TESTED framework—a six-phase approach that transforms traditional quality activities into hypothesis-driven scientific inquiry:
T – Target Definition
Traditional Approach: Identify potential quality risks through brainstorming or checklist methods. TESTED Approach: Define specific, measurable quality targets based on patient impact and process understanding. Each target should specify not just what we want to achieve, but why achieving it matters for patient safety and product efficacy.
E – Evidence Assembly
Traditional Approach: Collect available data to support predetermined conclusions. TESTED Approach: Systematically gather evidence from multiple sources—historical data, scientific literature, process knowledge, and regulatory guidance—without predetermined outcomes. This evidence serves as the foundation for hypothesis development rather than justification for existing practices.
S – Scientific Hypothesis Formation
Traditional Approach: Develop risk assessments based on expert judgment and generic failure modes. TESTED Approach: Formulate specific, falsifiable hypotheses about what drives quality outcomes. These hypotheses should make testable predictions about system behavior under different conditions.
T – Testing Design
Traditional Approach: Design validation studies to demonstrate compliance with predetermined acceptance criteria. TESTED Approach: Design experiments and monitoring systems to test hypothesis validity. Testing should be capable of falsifying hypotheses if they are incorrect, not just confirming predetermined expectations.
E – Evaluation and Analysis
Traditional Approach: Analyze results to demonstrate system adequacy. TESTED Approach: Rigorously evaluate evidence against hypothesis predictions. When hypotheses are falsified, this provides valuable information about system behavior rather than failure to be explained away.
D – Decision and Adaptation
Traditional Approach: Implement controls based on risk assessment outcomes. TESTED Approach: Adapt quality systems based on genuine learning about what drives quality outcomes. Use hypothesis testing results to refine understanding and improve system design.
Application Examples
Cleaning Validation Transformation
Traditional Approach: Demonstrate that cleaning procedures consistently achieve residue levels below acceptance criteria.
TESTED Implementation:
Target: Prevent cross-contamination between products while optimizing cleaning efficiency
Evidence: Historical contamination data, scientific literature on cleaning mechanisms, process capability data
Hypothesis: Contact time with cleaning solution above 12 minutes combined with mechanical action intensity >150 RPM will achieve >99.9% protein removal regardless of product sequence, with failure probability <1% when both parameters are maintained simultaneously
Testing: Designed experiments varying contact time and mechanical action across different product sequences
Evaluation: Results confirmed the importance of the interaction but revealed that product sequence affects required contact time by up to 40%
Decision: Revised cleaning procedure to account for product-specific requirements while maintaining hypothesis-driven monitoring
Process Control Strategy Development
Traditional Approach: Establish critical process parameters and control limits based on process validation studies.
TESTED Implementation:
Target: Ensure consistent product quality while enabling process optimization
Evidence: Process development data, literature on similar processes, regulatory precedents
Hypothesis: Product quality is primarily controlled by the interaction between temperature (±2°C) and pH (±0.1 units) during the reaction phase, with environmental factors contributing <5% to overall variability when these parameters are controlled
Testing: Systematic evaluation of parameter interactions using designed experiments
Evaluation: Temperature-pH interaction confirmed, but humidity found to have >10% impact under specific conditions
Decision: Enhanced control strategy incorporating environmental monitoring with hypothesis-based action limits
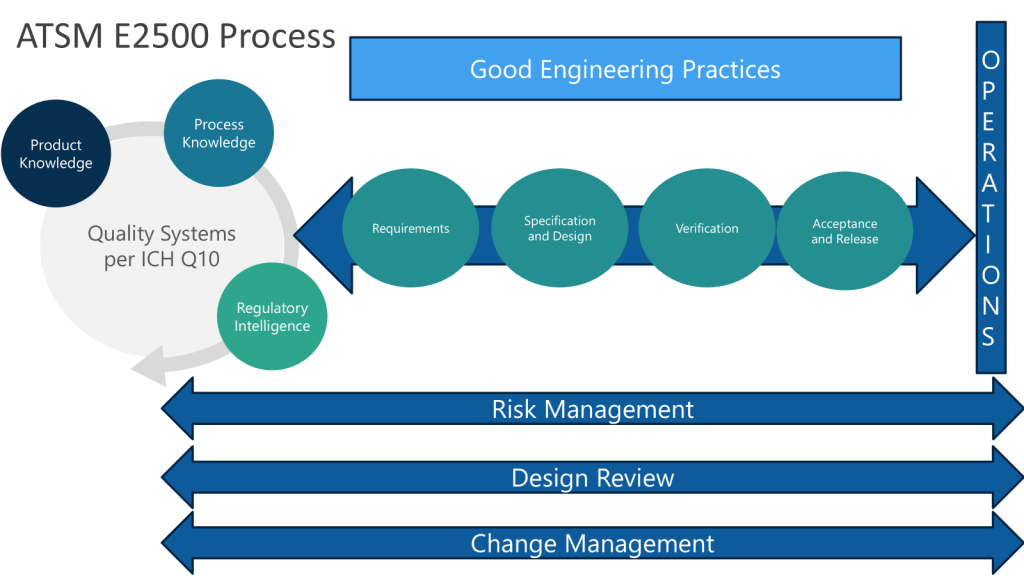
Traditional document management approaches, rooted in paper-based paradigms, create artificial boundaries between engineering activities and quality oversight. These silos become particularly problematic when implementing Quality Risk Management-based integrated Commissioning and Qualification strategies. The solution lies not in better document control procedures, but in embracing data-centric architectures that treat documents as dynamic views of underlying quality data rather than static containers of information.
The Engineering Quality Process: Beyond Document Control
The Engineering Quality Process (EQP) represents an evolution beyond traditional document management, establishing the critical interface between Good Engineering Practice and the Pharmaceutical Quality System. This integration becomes particularly crucial when we consider that engineering documents are not merely administrative artifacts—they are the embodiment of technical knowledge that directly impacts product quality and patient safety.
EQP implementation requires understanding that documents exist within complex data ecosystems where engineering specifications, risk assessments, change records, and validation protocols are interconnected through multiple quality processes. The challenge lies in creating systems that maintain this connectivity while ensuring ALCOA+ principles are embedded throughout the document lifecycle.
Building Systematic Document Governance
The foundation of effective GEP document management begins with recognizing that documents serve multiple masters—engineering teams need technical accuracy and accessibility, quality assurance requires compliance and traceability, and operations demands practical usability. This multiplicity of requirements necessitates what I call “multi-dimensional document governance”—systems that can simultaneously satisfy engineering, quality, and operational needs without creating redundant or conflicting documentation streams.
Effective governance structures must establish clear boundaries between engineering autonomy and quality oversight while ensuring seamless information flow across these interfaces. This requires moving beyond simple approval workflows toward sophisticated quality risk management integration where document criticality drives the level of oversight and control applied.
Electronic Quality Management System Integration: The Technical Architecture
The integration of eQMS platforms with engineering documentation can be surprisingly complex. The fundamental issue is that most eQMS solutions were designed around quality department workflows, while engineering documents flow through fundamentally different processes that emphasize technical iteration, collaborative development, and evolutionary refinement.
Core Integration Principles
Unified Data Models: Rather than treating engineering documents as separate entities, leading implementations create unified data models where engineering specifications, quality requirements, and validation protocols share common data structures. This approach eliminates the traditional handoffs between systems and creates seamless information flow from initial design through validation and into operational maintenance.
Risk-Driven Document Classification: We need to move beyond user driven classification and implement risk classification algorithms that automatically determine the level of quality oversight required based on document content, intended use, and potential impact on product quality. This automated classification reduces administrative burden while ensuring critical documents receive appropriate attention.
Contextual Access Controls: Advanced eQMS platforms provide dynamic permission systems that adjust access rights based on document lifecycle stage, user role, and current quality status. During active engineering development, technical teams have broader access rights, but as documents approach finalization and quality approval, access becomes more controlled and audited.
Validation Management System Integration
The integration of electronic Validation Management Systems (eVMS) represents a particularly sophisticated challenge because validation activities span the boundary between engineering development and quality assurance. Modern implementations create bidirectional data flows where engineering documents automatically populate validation protocols, while validation results feed back into engineering documentation and quality risk assessments.
Protocol Generation: Advanced systems can automatically generate validation protocols from engineering specifications, user requirements, and risk assessments. This automation ensures consistency between design intent and validation activities while reducing the manual effort typically required for protocol development.
Evidence Linking: Sophisticated eVMS platforms create automated linkages between engineering documents, validation protocols, execution records, and final reports. These linkages ensure complete traceability from initial requirements through final qualification while maintaining the data integrity principles essential for regulatory compliance.
Continuous Verification: Modern systems support continuous verification approaches aligned with ASTM E2500 principles, where validation becomes an ongoing process integrated with change management rather than discrete qualification events.
Data Integrity Foundations: ALCOA+ in Engineering Documentation
The application of ALCOA+ principles to engineering documentation can create challenges because engineering processes involve significant collaboration, iteration, and refinement—activities that can conflict with traditional interpretations of data integrity requirements. The solution lies in understanding that ALCOA+ principles must be applied contextually, with different requirements during active development versus finalized documentation.
Attributability in Collaborative Engineering
Engineering documents often represent collective intelligence rather than individual contributions. Address this challenge through granular attribution mechanisms that can track individual contributions to collaborative documents while maintaining overall document integrity. This includes sophisticated version control systems that maintain complete histories of who contributed what content, when changes were made, and why modifications were implemented.
Contemporaneous Recording in Design Evolution
Traditional interpretations of contemporaneous recording can conflict with engineering design processes that involve iterative refinement and retrospective analysis. Implement design evolution tracking that captures the timing and reasoning behind design decisions while allowing for the natural iteration cycles inherent in engineering development.
Managing Original Records in Digital Environments
The concept of “original” records becomes complex in engineering environments where documents evolve through multiple versions and iterations. Establish authoritative record concepts where the system maintains clear designation of authoritative versions while preserving complete historical records of all iterations and the reasoning behind changes.
Best Practices for eQMS Integration
Systematic Architecture Design
Effective eQMS integration begins with architectural thinking rather than tool selection. Organizations must first establish clear data models that define how engineering information flows through their quality ecosystem. This includes mapping the relationships between user requirements, functional specifications, design documents, risk assessments, validation protocols, and operational procedures.
Cross-Functional Integration Teams: Successful implementations establish integrated teams that include engineering, quality, IT, and operations representatives from project inception. These teams ensure that system design serves all stakeholders’ needs rather than optimizing for a single department’s workflows.
Phased Implementation Strategies: Rather than attempting wholesale system replacement, leading organizations implement phased approaches that gradually integrate engineering documentation with quality systems. This allows for learning and refinement while maintaining operational continuity.
Change Management Integration
The integration of change management across engineering and quality systems represents a critical success factor. Create unified change control processes where engineering changes automatically trigger appropriate quality assessments, risk evaluations, and validation impact analyses.
Automated Impact Assessment: Ensure your system can automatically assess the impact of engineering changes on existing validation status, quality risk profiles, and operational procedures. This automation ensures that changes are comprehensively evaluated while reducing the administrative burden on technical teams.
Stakeholder Notification Systems: Provide contextual notifications to relevant stakeholders based on change impact analysis. This ensures that quality, operations, and regulatory affairs teams are informed of changes that could affect their areas of responsibility.
Knowledge Management Integration
Capturing Engineering Intelligence
One of the most significant opportunities in modern GEP document management lies in systematically capturing engineering intelligence that traditionally exists only in informal networks and individual expertise. Implement knowledge harvesting mechanisms that can extract insights from engineering documents, design decisions, and problem-solving approaches.
Design Decision Rationale: Require and capture the reasoning behind engineering decisions, not just the decisions themselves. This creates valuable organizational knowledge that can inform future projects while providing the transparency required for quality oversight.
Lessons Learned Integration: Rather than maintaining separate lessons learned databases, integrate insights directly into engineering templates and standard documents. This ensures that organizational knowledge is immediately available to teams working on similar challenges.
Expert Knowledge Networks
Create dynamic expert networks where subject matter experts are automatically identified and connected based on document contributions, problem-solving history, and technical expertise areas. These networks facilitate knowledge transfer while ensuring that critical engineering knowledge doesn’t remain locked in individual experts’ experience.
Technology Platform Considerations
System Architecture Requirements
Effective GEP document management requires platform architectures that can support complex data relationships, sophisticated workflow management, and seamless integration with external engineering tools. This includes the ability to integrate with Computer-Aided Design systems, engineering calculation tools, and specialized pharmaceutical engineering software.
API Integration Capabilities: Modern implementations require robust API frameworks that enable integration with the diverse tool ecosystem typically used in pharmaceutical engineering. This includes everything from CAD systems to process simulation software to specialized validation tools.
Scalability Considerations: Pharmaceutical engineering projects can generate massive amounts of documentation, particularly during complex facility builds or major system implementations. Platforms must be designed to handle this scale while maintaining performance and usability.
Validation and Compliance Framework
The platforms supporting GEP document management must themselves be validated according to pharmaceutical industry standards. This creates unique challenges because engineering systems often require more flexibility than traditional quality management applications.
GAMP 5 Compliance: Follow GAMP 5 principles for computerized system validation while maintaining the flexibility required for engineering applications. This includes risk-based validation approaches that focus validation efforts on critical system functions.
Continuous Compliance: Modern systems support continuous compliance monitoring rather than point-in-time validation. This is particularly important for engineering systems that may receive frequent updates to support evolving project needs.
Building Organizational Maturity
Cultural Transformation Requirements
The successful implementation of integrated GEP document management requires cultural transformation that goes beyond technology deployment. Engineering organizations must embrace quality oversight as value-adding rather than bureaucratic, while quality organizations must understand and support the iterative nature of engineering development.
Cross-Functional Competency Development: Success requires developing transdisciplinary competence where engineering professionals understand quality requirements and quality professionals understand engineering processes. This shared understanding is essential for creating systems that serve both communities effectively.
Evidence-Based Decision Making: Organizations must cultivate cultures that value systematic evidence gathering and rigorous analysis across both technical and quality domains. This includes establishing standards for what constitutes adequate evidence for engineering decisions and quality assessments.
Maturity Model Implementation
Organizations can assess and develop their GEP document management capabilities using maturity model frameworks that provide clear progression paths from reactive document control to sophisticated knowledge-enabled quality systems.
Level 1 – Reactive: Basic document control with manual processes and limited integration between engineering and quality systems.
Level 2 – Developing: Electronic systems with basic workflow automation and beginning integration between engineering and quality processes.
Level 3 – Systematic: Comprehensive eQMS integration with risk-based document management and sophisticated workflow automation.
Level 4 – Integrated: Unified data architectures with seamless information flow between engineering, quality, and operational systems.
Level 5 – Optimizing: Knowledge-enabled systems with predictive analytics, automated intelligence extraction, and continuous improvement capabilities.
Future Directions and Emerging Technologies
Artificial Intelligence Integration
The convergence of AI technologies with GEP document management creates unprecedented opportunities for intelligent document analysis, automated compliance checking, and predictive quality insights. The promise is systems that can analyze engineering documents to identify potential quality risks, suggest appropriate validation strategies, and automatically generate compliance reports.
Natural Language Processing: AI-powered systems can analyze technical documents to extract key information, identify inconsistencies, and suggest improvements based on organizational knowledge and industry best practices.
Predictive Analytics: Advanced analytics can identify patterns in engineering decisions and their outcomes, providing insights that improve future project planning and risk management.
Building Excellence Through Integration
The transformation of GEP document management from compliance-driven bureaucracy to value-creating knowledge systems represents one of the most significant opportunities available to pharmaceutical organizations. Success requires moving beyond traditional document control paradigms toward data-centric architectures that treat documents as dynamic views of underlying quality data.
The integration of eQMS platforms with engineering workflows, when properly implemented, creates seamless quality ecosystems where engineering intelligence flows naturally through validation processes and into operational excellence. This integration eliminates the traditional handoffs and translation losses that have historically plagued pharmaceutical quality systems while maintaining the oversight and control required for regulatory compliance.
Organizations that embrace these integrated approaches will find themselves better positioned to implement Quality by Design principles, respond effectively to regulatory expectations for science-based quality systems, and build the organizational knowledge capabilities required for sustained competitive advantage in an increasingly complex regulatory environment.
The future belongs to organizations that can seamlessly blend engineering excellence with quality rigor through sophisticated information architectures that serve both engineering creativity and quality assurance requirements. The technology exists; the regulatory framework supports it; the question remaining is organizational commitment to the cultural and architectural transformations required for success.
As we continue evolving toward more evidence-based quality practice, the organizations that invest in building coherent, integrated document management systems will find themselves uniquely positioned to navigate the increasing complexity of pharmaceutical quality requirements while maintaining the engineering innovation essential for bringing life-saving products to market efficiently and safely.