The FDA does not publish many notices on data integrity, so folks should pay attention to yesterday’s publication on Synapse Labs Pvt. Ltd. (Synapse)—a contract research organization (CRO) based in Pune, India, and the need to repeat any studies conducted there.
Category: compliance
Section 711 of FDASIA and Regulatory Obligations
Too often, I see folks in pharma focus on 21 CFR Chapter 1, or at best all three chapters, maybe know the guidances and pay attention to little else. Unfortunately, that approach will often get one in trouble.
Section 711 of the Food and Drug Administration Safety and Innovation Act (FDASIA) amended the Federal Food, Drug, and Cosmetic Act (FD&C Act) to enhance the safety and quality of the drug supply chain. Specifically Section 711 amends Section 501(a)(2)(B) of the FD&C Act by adding the following sentence:
“For purposes of paragraph (a)(2)(B), the term ‘current good manufacturing practice’ includes the implementation of oversight and controls over the manufacture of drugs to ensure quality, including managing the risk of and establishing the safety of raw materials, materials used in the manufacturing of drugs, and finished drug products.”
This amendment clarifies that current good manufacturing practice (CGMP) requirements for drugs include:
- Implementing oversight and controls over the entire manufacturing process to ensure quality.
- Managing the risks related to raw materials, other materials used in manufacturing, and finished drug products to establish their safety.
In essence, Section 711 expands the FDA’s CGMP authority to explicitly cover supply chain management and drug manufacturers’ oversight of their suppliers and contract manufacturing operations. It also allows the FDA to enforce supply chain control requirements during inspections.
The legislative history shows that Congress intended to significantly expand the FDA’s authority over the increasingly global drug supply chain through this provision. It allows the FDA to scrutinize how manufacturers select, qualify, and oversee suppliers of raw materials and contract manufacturers to ensure drug quality and safety.
Please note that the FDA gets this expanded authority without revising 21CFR. That’s how it works; Congress can do that. Will we eventually see some 21 CFR updates? I have no idea.
But what this does mean is that the FDA has the authority to:
- Inspect risk management for GMPs, and assume you have it. What does good risk management look like? The agency has adopted ICH Q9(r1) as guidance, so start there.
- Inspect your supplier management, which includes qualifying and overseeing suppliers and contract manufacturers.
I’ve started to receive regulatory intelligence that this is coming up in inspections. Expect to be asked for the risk management evidence and for supplier qualification and oversight evidence.
Back Up and Recovery Testing
Backup and recovery testing are critical to ensuring data integrity and business continuity for critical computerized systems. They are also a hard regulatory requirement in our computer system lifecycle.
Part 11 (21 CFR 11.10 and 11.30) requires that:
“For the availability of computerized systems supporting critical processes, provisions should be made to ensure continuity of the systems in the event of an incident or system failure. This includes implementing adequate backup and recovery measures, as well as providing sufficient system redundancy and failover mechanisms.”
Part 11 also requires that “The backup and recovery processes must be validated in order to ensure that they operate in an effective and reliable manner.”
Similarly, Annex 11 requires that backup and recovery processes be validated to ensure they operate reliably and effectively. Annex 11 also requires that the validation process be documented and includes a risk assessment of the system’s critical processes.
Similar requirements can be found across the GxP data integrity requirements.
The regulatory requirements require that backup and recovery processes be validated to ensure they can reliably recover the system in case of an incident or failure. This validation process must be documented, including a risk assessment of the system’s critical processes.
Backup and recovery testing:
- Verifies Backup Integrity: Testing backups lets you verify that the backup data is complete, accurate, and not corrupted. It ensures that the backed-up data can be reliably restored when needed, maintaining the integrity of the original data.
- Validates Recovery Procedures: Regularly testing the recovery process helps identify and resolve any issues or gaps in the recovery procedures. This ensures that the data can be restored wholly and correctly, preserving its integrity during recovery.
- Identifies Data Corruption: Testing can reveal data corruption that may have gone unnoticed. By restoring backups and comparing them with the original data, you can detect and address any data integrity issues before they become critical.
- Improves Disaster Preparedness: Regular backup and recovery testing helps organizations identify and address potential issues before a disaster strikes. This improves the organization’s preparedness and ability to recover data with integrity in a disaster or data loss incident.
- Maintains Business Continuity: Backup and recovery testing helps maintain business continuity by ensuring that backups are reliable and recovery procedures are adequate. Organizations can minimize downtime and data loss, ensuring the integrity of critical business data and operations.
To maintain data integrity, it is recommended that backup and recovery testing be performed regularly. This should follow industry best practices and adhere to the organization’s recovery time objectives (RTOs) and recovery point objectives (RPOs). Testing should cover various scenarios, including full system restores, partial data restores, and data validation checks.
| Level | Description | Key Activities | Frequency |
| Backup Tests | Ensures data is backed up correctly and consistently. | – Check backup infrastructure health – Verify data consistency – Ensure all critical data is covered – Check security settings | Regularly (daily, weekly, monthly) |
| Recovery Tests | Ensures data can be restored effectively and within required timeframes. | – Test recovery time and point objectives (RTO and RPO) – Define and test various recovery scopes – Schedule tests to avoid business disruption – Document all tests and results | Regularly (quarterly, biannually, annually) |
| Disaster Recovery Tests | Ensures the disaster recovery plan is effective and feasible. | – Perform disaster recovery scenarios – Test failover and failback operations – Coordinate with all relevant teams and stakeholders | Less frequent (once or twice a year) |
By incorporating backup and recovery testing into the data lifecycle, organizations can have confidence in their ability to recover data with integrity, minimizing the risk of data loss or corruption and ensuring business continuity in the face of disasters or data loss incidents.
| Aspect | Backup Tests | Recovery Tests |
| Objective | Verify data integrity and backup processes | Ensure data and systems can be successfully restored |
| Focus | Data backup and storage | Comprehensive recovery of data, applications, and infrastructure |
| Processes | Data copy verification, consistency checks, storage verification | Full system restore, spot-checking, disaster simulation |
| Scope | Data-focused | Broader scope including systems and infrastructure |
| Frequency | Regular intervals (daily, weekly, monthly) | Less frequent but more thorough |
| Testing Areas | Backup scheduling, data transfer, storage capacity | Recovery time objectives (RTO), recovery point objectives (RPO), failover/failback |
| Validation | Backup data is complete and accessible | Restored data and systems are fully functional |
Batch and the Batch Record
Inevitably, in biotech, with our manufacturing processes such as cell culture, fermentation, and purification, we ask the question (especially with continuous manufacturing), “Just what is a batch anyway.” Luckily for us, the ISA S88.01 provides a standard, with models and terminology, to give us a structured framework to define, control, and automate batch processes effectively
ISA S88.01 (ANSI/ISA-88) standardizes batch control terminology by providing a consistent set of models and terminology for describing all the aspects of batch processing. This standardization helps improve communication between all parties involved in batch control, including users, vendors, and engineers.
- Models and Terminology: ISA S88.01 defines a set of models and terminology to describe batch control’s physical and procedural aspects. This includes the physical model, which outlines the hierarchical structure of equipment, and the procedural control model, which details the sequence of operations and phases involved in batch processing.
- Physical Model: The physical model begins at the enterprise level and includes sites, areas, process cells, units, equipment, and control modules. This hierarchical structure ensures that all physical components involved in batch processing are consistently described.
- Procedural Control Model: This model consists of recipe procedures, unit procedures, operations, and phases. Each level in this hierarchy represents a different level of detail in the batch process, from high-level procedures to specific actions performed by equipment.
- Recipe Types and Contents: ISA S88.01 standardizes the types of recipes (general, site, master, and control) and their contents, which include the header, formula, equipment requirements, procedure, and other necessary information. This ensures recipes are consistently structured and understood across different systems and organizations.
- State Definitions: The standard defines various states that units or phases can transition through during their operation, such as idle, running, held, paused, aborted, and completed. These states provide a standardized framework for interaction between recipe phases and control system equipment.
- Data Structures and Guidelines: ISA S88.01 provides guidelines for data structures and batch control languages, simplifying programming, configuration tasks, and communication between system components. This helps ensure that data is consistently managed and communicated within the batch control system.
The Batch Record
Batch records are the primary documentation that captures the real-time performance of production records. Batch records are crucial to confirming that all expected and required actions have been completed within parameters to produce a product that meets specifications and complies with quality standards.
The Master Batch Record (MBR) is the version-controlled documentation necessary to trace the complete cycle of manufacture of a batch of product, from the dispensing of materials through all processing, testing, and subsequent packaging to the dispatch for sale or supply of the finished product. This documentation includes quality control, quality assurance, and environmental data relevant to the intended manufacturing.
The MBR may be segmented on intended manufacturing and testing stages, each part controlled separately.
The Production Batch Record (PBR) is issued for manufacturing one (or more) batches from the MBR and is compiled during manufacturing.
The MBR and PBR may be controlled in the document management system, within a manufacturing execution system (MES)/electronic batch record (EBR) platform, or some hybrid. Parts may also be found within the LIMS, data historian, and other electronic systems. A critical part of building the MBR is ensuring the correct connections between it and data in specific electronic platforms.
| Electronic System | Description | |
| Master Production Record | Master Recipe | Contains product name or designation, recipe designation or version, formulas, equipment requirements or classes, sequence of activities, procedures, normalized bill of materials (quantity per unit volume to produce) |
| Work Instructions | Additional detailed instructions – may include electronic SOPs or SOP references | |
| Critical Process Parameters | Required Process Parameters that are to be checked or monitored or are to be downloaded to other systems such as automation | |
| Production Batch Record | Control Recipe | A Master Recipe dispatched or otherwise made available in manufacturing-related areas for Execution. Includes Master Recipe information with the addition of schedule, specific quantity to make, actual target bill of materials quantities, and other data for the batch and production instance |
| Electronic Production Record | A store of data and information created by systems or entered by personnel during execution of Control Recipes May be located in one or more systems or databases Data may or may not be stored in human readable format | |
| Production Report | Data and information in human-readable format, presented either in electronic or paper format for activities such as review, disposition, investigation, audit, and analysis. |
FDA CY 2023 Shortage Report
The FDA has published its calendar year 2023 Shortage Report to Congress. Surprising no one, shortages continue to be an issue.
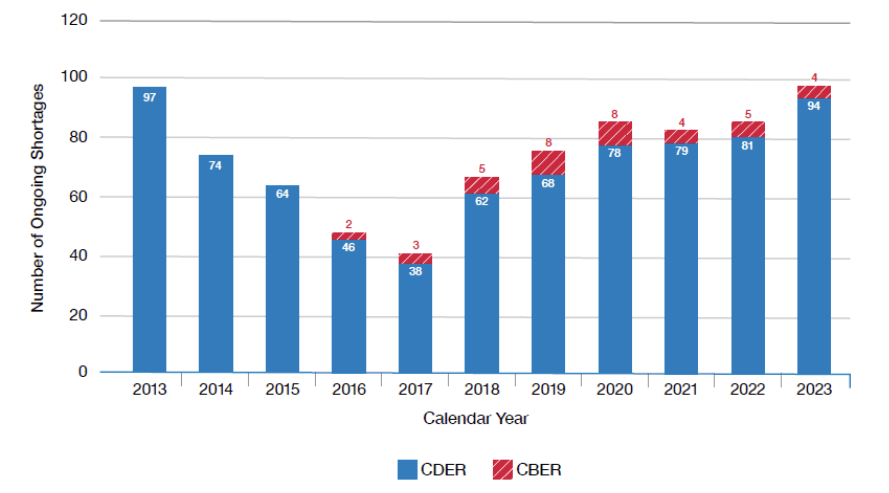
Section 5 is the fun part, highlighting continued efforts for QMM, collaboration with foreign regulatory bodies, and other activities.
