We have an unfortunate habit of conflating regulatory process requirements with specific system functionality requirements. This confusion manifests most perversely in User Requirements Specifications that contain nebulous statements like “the system shall comply with 21 CFR Part 11” or “the system must meet EU GMP Annex 11 requirements.” These high-level regulatory citations represent a fundamental misunderstanding of what user requirements should accomplish and demonstrate a dangerous abdication of the detailed thinking required for effective validation.
The core problem is simple yet profound: lifecycle, risk management, and validation are organizational processes, not system characteristics. When we embed these process-level concepts into system requirements, we create validation exercises that test compliance theater rather than functional reality.
The Distinction That Changes Everything
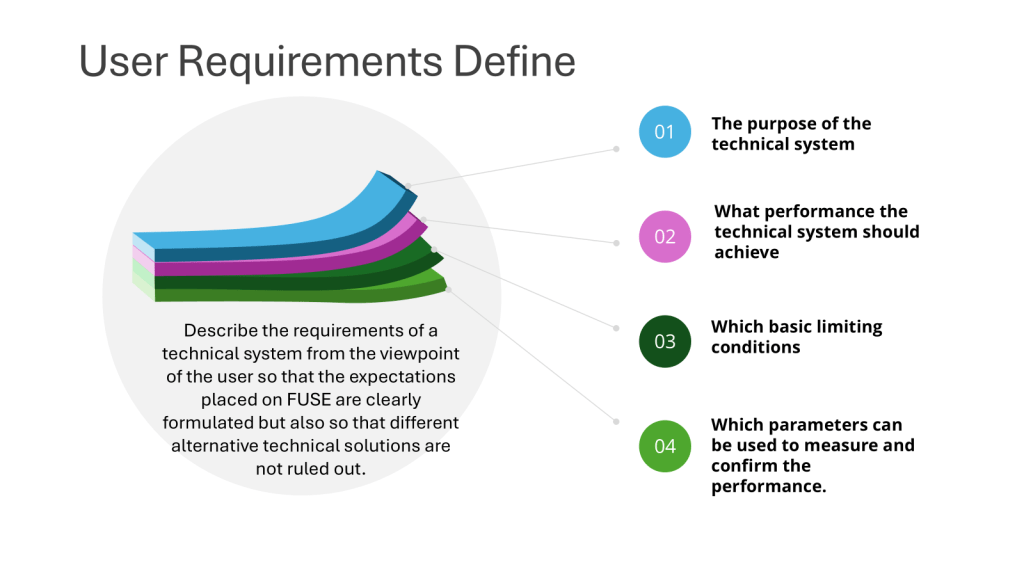
User requirements specifications serve as the foundational document identifying what a system must do to meet specific business needs, product requirements, and operational constraints. They translate high-level business objectives into measurable, testable, and verifiable system behaviors. User requirements focus on what the system must accomplish, not how the organization manages its regulatory obligations.
Consider the fundamental difference between these approaches:
Problematic High-Level Requirement: “The system shall comply with 21 CFR Part 11 validation requirements.”
Proper Detailed Requirements:
“The system shall generate time-stamped audit trails for all data modifications, including user ID, date/time, old value, new value, and reason for change”
“The system shall enforce unique user identification through username/password combinations with minimum 8-character complexity requirements”
“The system shall prevent deletion of electronic records while maintaining complete audit trail visibility”
“The system shall provide electronic signature functionality that captures the printed name, date/time, and meaning of the signature”
The problematic version tells us nothing about what the system actually needs to do. The detailed versions provide clear, testable criteria that directly support Part 11 compliance while specifying actual system functionality.
Process vs. System: Understanding the Fundamental Categories
Lifecycle management, risk assessment, and validation represent organizational processes that exist independently of any specific system implementation. These processes define how an organization approaches system development, operation, and maintenance—they are not attributes that can be “built into” software.
Lifecycle processes encompass the entire journey from initial system conception through retirement, including stages such as requirements definition, design, development, testing, deployment, operation, and eventual decommissioning. A lifecycle approach ensures systematic progression through these stages with appropriate documentation, review points, and decision criteria. However, lifecycle management cannot be embedded as a system requirement because it describes the framework within which system development occurs, not system functionality itself.
Risk management processes involve systematic identification, assessment, and mitigation of potential hazards throughout system development and operation. Risk management influences system design decisions and validation approaches, but risk management itself is not a system capability—it is an organizational methodology for making informed decisions about system requirements and controls.
Validation processes establish documented evidence that systems consistently perform as intended and meet all specified requirements. Validation involves planning, execution, and documentation of testing activities, but validation is something done to systems, not something systems possess as an inherent characteristic.
The Illusion of Compliance Through Citation
When user requirements specifications contain broad regulatory citations rather than specific functional requirements, they create several critical problems that undermine effective validation:
Untestable Requirements: How does one verify that a system “complies with Part 11”? Such requirements provide no measurable criteria, no specific behaviors to test, and no clear success/failure conditions. Verification becomes a subjective exercise in regulatory interpretation rather than objective measurement of system performance.
Validation Theater: Broad compliance statements encourage checkbox validation exercises where teams demonstrate regulatory awareness without proving functional capability. These validations often consist of mapping system features to regulatory sections rather than demonstrating that specific user needs are met.
Scope Ambiguity: Part 11 and Annex 11 contain numerous requirements, many of which may not apply to specific systems or use cases. Blanket compliance statements fail to identify which specific regulatory requirements are relevant and which system functions address those requirements.
Change Management Nightmares: When requirements reference entire regulatory frameworks rather than specific system behaviors, any regulatory update potentially impacts system validation status. This creates unnecessary re-validation burdens and regulatory uncertainty.
Building Requirements That Actually Work
Effective user requirements specifications address regulatory compliance through detailed, system-specific functional requirements that directly support regulatory objectives. This approach ensures that validation activities test actual system capabilities rather than regulatory awareness
Focus on Critical Quality Attributes: Rather than citing broad compliance frameworks, identify the specific product and process attributes that regulatory requirements are designed to protect. For pharmaceutical systems, this might include data integrity, product traceability, batch genealogy, or contamination prevention.
Translate Regulatory Intent into System Functions: Understand what each applicable regulation is trying to achieve, then specify system behaviors that accomplish those objectives. Part 11’s audit trail requirements, for example, aim to ensure data integrity and accountability—translate this into specific system capabilities for logging, storing, and retrieving change records.
Maintain Regulatory Traceability: Document the relationship between specific system requirements and regulatory drivers, but do so through traceability matrices or design rationale documents rather than within the requirements themselves. This maintains clear regulatory justification while keeping requirements focused on system functionality.
Enable Risk-Based Validation: Detailed functional requirements support risk-based validation approaches by clearly identifying which system functions are critical to product quality, patient safety, or data integrity. This enables validation resources to focus on genuinely important capabilities rather than comprehensive regulatory coverage.
The Process-System Interface: Getting It Right
The relationship between organizational processes and system requirements should be managed through careful analysis and translation, not through broad regulatory citations. Effective user requirements development involves several critical steps:
Process Analysis: Begin by understanding the organizational processes that the system must support. This includes manufacturing processes, quality control workflows, regulatory reporting requirements, and compliance verification activities. However, the focus should be on what the system must enable, not how the organization manages compliance.
Regulatory Gap Analysis: Identify specific regulatory requirements that apply to the intended system use. Analyze these requirements to understand their functional implications for system design, but avoid copying regulatory language directly into system requirements.
Functional Translation: Convert regulatory requirements into specific, measurable system behaviors. This translation process requires deep understanding of both regulatory intent and system capabilities, but produces requirements that can be objectively verified.
Organizational Boundary Management: Clearly distinguish between requirements for system functionality and requirements for organizational processes. System requirements should focus exclusively on what the technology must accomplish, while process requirements address how the organization will use, maintain, and govern that technology.
Real-World Consequences of the Current Approach
The practice of embedding high-level regulatory requirements in user requirements specifications has created systemic problems throughout the pharmaceutical industry:
Validation Inefficiency: Teams spend enormous resources demonstrating broad regulatory compliance rather than proving that systems meet specific user needs. This misallocation of validation effort undermines both regulatory compliance and system effectiveness.
Inspection Vulnerability: When regulatory inspectors evaluate systems against broad compliance claims, they often identify gaps between high-level assertions and specific system capabilities. Detailed functional requirements provide much stronger inspection support by demonstrating specific regulatory compliance mechanisms.
System Modification Complexity: Changes to systems with broad regulatory requirements often trigger extensive re-validation activities, even when the changes don’t impact regulatory compliance. Specific functional requirements enable more targeted change impact assessments.
Cross-Functional Confusion: Development teams, validation engineers, and quality professionals often interpret broad regulatory requirements differently, leading to inconsistent implementation and validation approaches. Detailed functional requirements provide common understanding and clear success criteria.
A Path Forward: Detailed Requirements for Regulatory Success
The solution requires fundamental changes in how the pharmaceutical industry approaches user requirements development and regulatory compliance documentation:
Separate Compliance Strategy from System Requirements: Develop comprehensive regulatory compliance strategies that identify applicable requirements and define organizational approaches for meeting them, but keep these strategies distinct from system functional requirements. Use the compliance strategy to inform requirements development, not replace it.
Invest in Requirements Translation: Build organizational capability for translating regulatory requirements into specific, testable system functions. This requires regulatory expertise, system knowledge, and requirements engineering skills working together.
Implement Traceability Without Embedding: Maintain clear traceability between system requirements and regulatory drivers through external documentation rather than embedded citations. This preserves regulatory justification while keeping requirements focused on system functionality.
Focus Validation on Function: Design validation approaches that test system capabilities directly rather than compliance assertions. This produces stronger regulatory evidence while ensuring system effectiveness.
Lifecycle, risk management, and validation are organizational processes that guide how we develop and maintain systems—they are not system requirements themselves. When we treat them as such, we undermine both regulatory compliance and system effectiveness. The time has come to abandon this regulatory red herring and embrace requirements practices worthy of the products and patients we serve.
Writing user requirements that simply state “the system shall meet FDA 21 CFR Part 11 requirements” or “the system shall comply with EU Annex 11” is fundamentally bad practice. These high-level regulatory statements create ambiguity, shift responsibility inappropriately, and fail to provide the specific, testable criteria that effective requirements demand.
The Core Problem: Regulatory References Aren’t Technical Requirements
User requirements must be specific, measurable, and testable. When we write “the system shall meet Annex 11 and Part 11 requirements,” we’re not writing a technical requirement at all—we’re writing a reference to a collection of regulatory provisions that may or may not apply to our specific system context. This creates several fundamental problems that undermine the entire validation and verification process.
The most critical issue is ambiguity of technical scope. Annex 11 and Part 11 contains numerous provisions, but not all apply to every system. Some provisions address closed systems, others open systems. Some apply only when electronic records replace paper records, others when organizations rely on electronic records to perform regulated activities. Without specifying which technical provisions apply and how they translate into system functionality, we leave interpretation to individual team members—a recipe for inconsistent implementation.
Technical verification becomes impossible with such high-level statements. How does a tester verify that a system “meets Part 11”? They would need to become regulatory experts, interpret which provisions apply, translate those into testable criteria, and then execute tests—work that should have been done during requirements definition. This shifting of analytical responsibility from requirements authors to testers violates fundamental engineering principles.
Why This Happens: The Path of Least Resistance
The temptation to write high-level regulatory requirements stems from several understandable but misguided motivations. Requirements authors often lack deep regulatory knowledge and find it easier to reference entire regulations rather than analyze which specific technical provisions apply to their system. This approach appears comprehensive while avoiding the detailed work of regulatory interpretation.
Time pressure exacerbates this tendency. Writing “meet Part 11” takes minutes; properly analyzing regulatory requirements, determining technical applicability, and translating them into specific, testable statements takes days or weeks. Under project pressure, teams often choose the quick path without considering downstream consequences.
There’s also a false sense of completeness. Referencing entire regulations gives the impression of thorough coverage when it actually provides no technical coverage at all. It’s the requirements equivalent of writing “the system shall work properly”, technically correct but utterly useless for implementation or testing purposes.
Better Approach: Technical User Requirements
Effective regulatory user requirements break down high-level regulatory concepts into specific, measurable technical statements that directly address system functionality. Rather than saying “meet Part 11,” we need requirements that specify exactly what the system must do technically.
Access Control Requirements
Instead of: “The system shall meet Part 11 access control requirements”
Write:
“The system shall authenticate users through unique user ID and password combinations, where each combination is assigned to only one individual”
“The system shall automatically lock user sessions after 30 minutes of inactivity and require re-authentication”
“The system shall maintain an electronic log of all user authentication attempts, including failed attempts, with timestamps and user identifiers”
Electronic Record Generation Requirements
Instead of: “The system shall generate Part 11 compliant electronic records”
Write:
“The system shall generate electronic records that include all data required by the predicate rule, with no omission of required information”
“The system shall time-stamp all electronic records using computer-generated timestamps that cannot be altered by system users”
“The system shall detect and flag any unauthorized alterations to electronic records through checksum validation”
Audit Trail Requirements
Instead of: “The system shall maintain Part 11 compliant audit trails”
Write:
“The system shall automatically record the user ID, date, time, and type of action for all data creation, modification, and deletion operations”
“The system shall store audit trail records in a format that prevents user modification or deletion”
“The system shall provide audit trail search and filter capabilities by user, date range, and record type”
Electronic Signature Requirements
Instead of: “The system shall support Part 11 electronic signatures”
Write:
“Electronic signature records shall include the signer’s printed name, date and time of signing, and the purpose of the signature”
“The system shall verify signer identity through authentication requiring both user ID and password before accepting electronic signatures“
“The system shall cryptographically link electronic signatures to their associated records to prevent signature transfer or copying”
Annex 11 Technical Examples
EU Annex 11 requires similar technical specificity but with some European-specific nuances. Here are better technical requirement examples:
System Security Requirements
Instead of: “The system shall meet Annex 11 security requirements”
Write:
“The system shall implement role-based access control where user privileges are assigned based on documented job responsibilities”
“The system shall encrypt all data transmission between system components using AES 256-bit encryption”
“The system shall maintain user session logs that record login time, logout time, and all system functions accessed during each session”
Data Integrity Requirements
Instead of: “The system shall ensure Annex 11 data integrity”
Write:
“The system shall implement automated backup procedures that create complete system backups daily and verify backup integrity”
“The system shall prevent simultaneous modification of the same record by multiple users through record locking mechanisms”
“The system shall maintain original raw data in unalterable format while allowing authorized users to add comments or corrections with full audit trails”
System Change Control Requirements
Instead of: “The system shall implement Annex 11 change control”
Write:
“The system shall require authorized approval through electronic workflow before implementing any configuration changes that affect GMP functionality”
“The system shall maintain a complete history of all system configuration changes including change rationale, approval records, and implementation dates”
“The system shall provide the ability to revert system configuration to any previous approved state through documented rollback procedures”
The Business Case for Technical Requirements
Technical requirements save time and money. While writing detailed requirements requires more upfront effort, it prevents costly downstream problems. Clear technical requirements eliminate the need for interpretation during design, reduce testing iterations, and prevent regulatory findings during inspections.
Technical traceability becomes meaningful when requirements are specific. We can trace from business needs through technical specifications to test cases and validation results. This traceability is essential for regulatory compliance and change control.
System quality improves systematically when everyone understands exactly what technical functionality needs to be built and tested. Vague requirements lead to assumption-driven development where different team members make different assumptions about what’s technically needed.
Implementation Strategy for Technical Requirements
Start by conducting regulatory requirement analysis as a separate technical activity before writing user requirements. Identify which regulatory provisions apply to your specific system and translate them into technical functionality. Document this analysis and use it as the foundation for technical requirement writing.
Engage both regulatory and technical experts early in the requirements process. Don’t expect requirements authors to become overnight regulatory experts, but do ensure they have access to both regulatory knowledge and technical understanding when translating regulatory concepts into system requirements.
Use technical requirement templates that capture the essential technical elements of common regulatory requirements. This ensures consistency across projects and reduces the analytical burden on individual requirements authors.
Review requirements for technical testability. Every requirement should have an obvious technical verification method. If you can’t immediately see how to test a requirement technically, it needs to be rewritten.
Technical Requirements That Actually Work
High-level regulatory references have no place in technical user requirements documents. They create technical ambiguity where clarity is needed, shift analytical work to inappropriate roles, and fail to provide the specific technical guidance necessary for successful system implementation.
Better technical requirements translate regulatory concepts into specific, measurable, testable statements that directly address system technical functionality. This approach requires more upfront effort but delivers better technical outcomes: clearer system designs, more efficient testing, stronger regulatory compliance, and systems that actually meet user technical needs.
The pharmaceutical industry has matured beyond accepting “it must be compliant” as adequate technical guidance. Our technical requirements must mature as well, providing the specific, actionable technical direction that modern development teams need to build quality systems that truly serve patients and regulatory expectations.
As I’ve emphasized in previous posts about crafting good user requirements and building FUSE(P) user requirements, technical specificity and testability remain the hallmarks of effective requirements writing. Regulatory compliance requirements demand this same technical rigor—perhaps more so, given the patient safety implications of getting the technical implementation wrong.
The pharmaceutical industry has operated for over a decade under the comfortable assumption that GAMP 5’s risk-based guidance for system requirements represented industry best practice—helpful, comprehensive, but ultimately voluntary. Section 6 of the draft Annex 11 moves many things from recommended to mandated. What GAMP 5 suggested as scalable guidance, Annex 11 codifies as enforceable regulation. For computer system validation professionals, this isn’t just an update—it’s a fundamental shift from “how we should do it” to “how we must do it.”
This transformation carries profound implications that extend far beyond documentation requirements. Section 6 represents the regulatory codification of modern system engineering practices, forcing organizations to abandon the shortcuts, compromises, and “good enough” approaches that have persisted despite GAMP 5’s guidance. More significantly, it establishes system requirements as the immutable foundation of validation rather than merely an input to the process.
For CSV experts who have spent years evangelizing GAMP 5 principles within organizations that treated requirements as optional documentation, Section 6 provides regulatory teeth that will finally compel comprehensive implementation. However, it also raises the stakes dramatically—what was once best practice guidance subject to interpretation becomes regulatory obligation subject to inspection.
The Mandatory Transformation: From Guidance to Regulation
6.1: GMP Functionality—The End of Requirements Optionality
The opening requirement of Section 6 eliminates any ambiguity about system requirements documentation: “A regulated user should establish and approve a set of system requirements (e.g. a User Requirements Specification, URS), which accurately describe the functionality the regulated user has automated and is relying on when performing GMP activities.”
This language transforms what GAMP 5 positioned as risk-based guidance into regulatory mandate. The phrase “should establish and approve” in regulatory context carries the force of “must“—there is no longer discretion about whether to document system requirements. Every computerized system touching GMP activities requires formal requirements documentation, regardless of system complexity, development approach, or organizational preference.
The scope is deliberately comprehensive, explicitly covering “whether a system is developed in-house, is a commercial off-the-shelf product, or is provided as-a-service” and “independently on whether it is developed following a linear or iterative software development process.” This eliminates common industry escapes: cloud services can’t claim exemption because they’re external; agile development can’t avoid documentation because it’s iterative; COTS systems can’t rely solely on vendor documentation because they’re pre-built.
The requirement for accuracy in describing “functionality the regulated user has automated and is relying on” establishes a direct link between system capabilities and GMP dependencies. Organizations must explicitly identify and document what GMP activities depend on system functionality, creating traceability between business processes and technical capabilities that many current validation approaches lack.
Major Strike Against the Concept of “Indirect”
The new draft Annex 11 explicitly broadens the scope of requirements for user requirements specifications (URS) and validation to cover all computerized systems with GMP relevance—not just those with direct product or decision-making impact, but also indirect GMP systems. This means systems that play a supporting or enabling role in GMP activities (such as underlying IT infrastructure, databases, cloud services, SaaS platforms, integrated interfaces, and any outsourced or vendor-managed digital environments) are fully in scope.
Section 6 of the draft states that user requirements must “accurately describe the functionality the regulated user has automated and is relying on when performing GMP activities,” with no exemption or narrower definition for indirect systems. It emphasizes that this principle applies “regardless of whether a system is developed in-house, is a commercial off-the-shelf product, or is provided as-a-service, and independently of whether it is developed following a linear or iterative software development process.” The regulated user is responsible for approving, controlling, and maintaining these requirements over the system’s lifecycle—even if the system is managed by a third party or only indirectly involved in GMP data or decision workflows.
Importantly, the language and supporting commentaries make it clear that traceability of user requirements throughout the lifecycle is mandatory for all systems with GMP impact—direct or indirect. There is no explicit exemption in the draft for indirect GMP systems. Regulatory and industry analyses confirm that the burden of documented, risk-assessed, and lifecycle-maintained user requirements sits equally with indirect systems as with direct ones, as long as they play a role in assuring product quality, patient safety, or data integrity.
In practice, this means organizations must extend their URS, specification, and validation controls to any computerized system that through integration, support, or data processing could influence GMP compliance. The regulated company remains responsible for oversight, traceability, and quality management of those systems, whether or not they are operated by a vendor or IT provider. This is a significant expansion from previous regulatory expectations and must be factored into computerized system inventories, risk assessments, and validation strategies going forward.
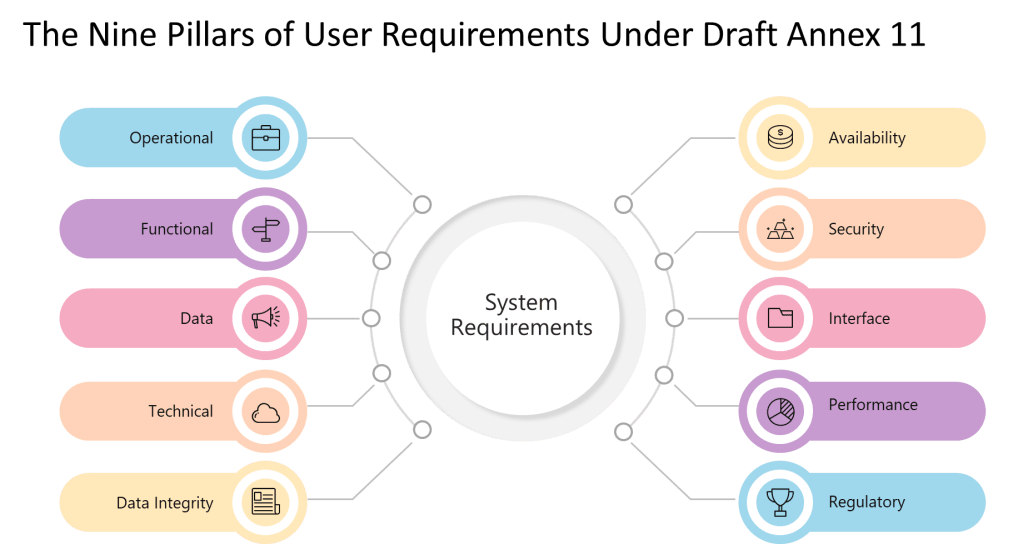
9 Pillars of a User Requirements
Pillar
Description
Practical Examples
Operational
Requirements describing how users will operate the system for GMP tasks.
Workflow steps, user roles, batch record creation.
Functional
Features and functions the system must perform to support GMP processes.
Explicit requirements imposed by GMP regulations and standards.
Part 11/Annex 11 compliance, data retention, auditability.
6.2: Extent and Detail—Risk-Based Rigor, Not Risk-Based Avoidance
Section 6.2 appears to maintain GAMP 5’s risk-based philosophy by requiring that “extent and detail of defined requirements should be commensurate with the risk, complexity and novelty of a system.” However, the subsequent specifications reveal a much more prescriptive approach than traditional risk-based frameworks.
The requirement that descriptions be “sufficient to support subsequent risk analysis, specification, design, purchase, configuration, qualification and validation” establishes requirements documentation as the foundation for the entire system lifecycle. This moves beyond GAMP 5’s emphasis on requirements as input to validation toward positioning requirements as the definitive specification against which all downstream activities are measured.
The explicit enumeration of requirement types—”operational, functional, data integrity, technical, interface, performance, availability, security, and regulatory requirements”—represents a significant departure from GAMP 5’s more flexible categorization. Where GAMP 5 allows organizations to define requirement categories based on system characteristics and business needs, Annex 11 mandates coverage of nine specific areas regardless of system type or risk level.
This prescriptive approach reflects regulatory recognition that organizations have historically used “risk-based” as justification for inadequate requirements documentation. By specifying minimum coverage areas, Section 6 establishes a floor below which requirements documentation cannot fall, regardless of risk assessment outcomes.
The inclusion of “process maps and data flow diagrams” as recommended content acknowledges the reality that modern pharmaceutical operations involve complex, interconnected systems where understanding data flows and process dependencies is essential for effective validation. This requirement will force organizations to develop system-level understanding rather than treating validation as isolated technical testing.
6.3: Ownership—User Accountability in the Cloud Era
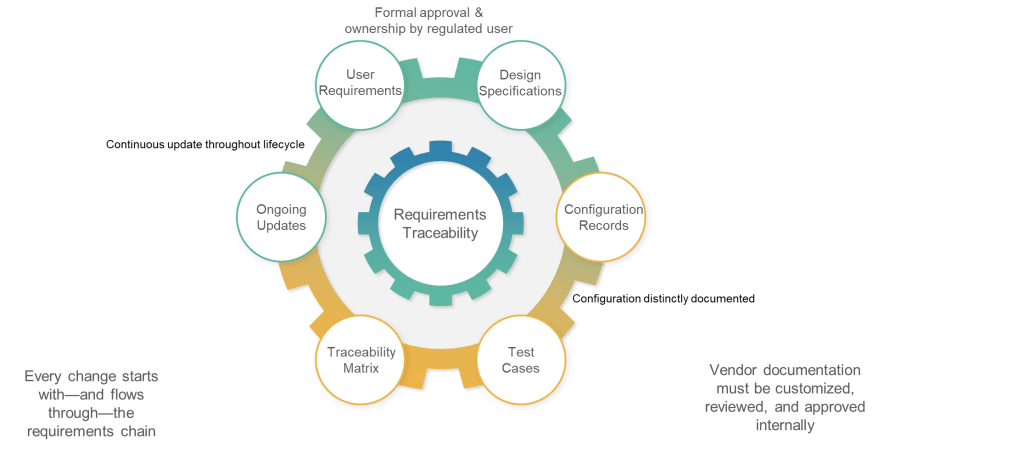
Perhaps the most significant departure from traditional industry practice, Section 6.3 addresses the growing trend toward cloud services and vendor-supplied systems by establishing unambiguous user accountability for requirements documentation. The requirement that “the regulated user should take ownership of the document covering the implemented version of the system and formally approve and control it” eliminates common practices where organizations rely entirely on vendor-provided documentation.
This requirement acknowledges that vendor-supplied requirements specifications rarely align perfectly with specific organizational needs, GMP processes, or regulatory expectations. While vendors may provide generic requirements documentation suitable for broad market applications, pharmaceutical organizations must customize, supplement, and formally adopt these requirements to reflect their specific implementation and GMP dependencies.
The language “carefully review and approve the document and consider whether the system fulfils GMP requirements and company processes as is, or whether it should be configured or customised” requires active evaluation rather than passive acceptance. Organizations cannot simply accept vendor documentation as sufficient—they must demonstrate that they have evaluated system capabilities against their specific GMP needs and either confirmed alignment or documented necessary modifications.
This ownership requirement will prove challenging for organizations using large cloud platforms or SaaS solutions where vendors resist customization of standard documentation. However, the regulatory expectation is clear: pharmaceutical companies cannot outsource responsibility for demonstrating that system capabilities meet their specific GMP requirements.
6.4: Update—Living Documentation, Not Static Archives
Section 6.4 addresses one of the most persistent failures in current validation practice: requirements documentation that becomes obsolete immediately after initial validation. The requirement that “requirements should be updated and maintained throughout the lifecycle of a system” and that “updated requirements should form the very basis for qualification and validation” establishes requirements as living documentation rather than historical artifacts.
This approach reflects the reality that modern computerized systems undergo continuous change through software updates, configuration modifications, hardware refreshes, and process improvements. Traditional validation approaches that treat requirements as fixed specifications become increasingly disconnected from operational reality as systems evolve.
The phrase “form the very basis for qualification and validation” positions requirements documentation as the definitive specification against which system performance is measured throughout the lifecycle. This means that any system change must be evaluated against current requirements, and any requirements change must trigger appropriate validation activities.
This requirement will force organizations to establish requirements management processes that rival those used in traditional software development organizations. Requirements changes must be controlled, evaluated for impact, and reflected in validation documentation—capabilities that many pharmaceutical organizations currently lack.
6.5: Traceability—Engineering Discipline for Validation
The traceability requirement in Section 6.5 codifies what GAMP 5 has long recommended: “Documented traceability between individual requirements, underlaying design specifications and corresponding qualification and validation test cases should be established and maintained.” However, the regulatory context transforms this from validation best practice to compliance obligation.
The emphasis on “effective tools to capture and hold requirements and facilitate the traceability” acknowledges that manual traceability management becomes impractical for complex systems with hundreds or thousands of requirements. This requirement will drive adoption of requirements management tools and validation platforms that can maintain automated traceability throughout the system lifecycle.
Traceability serves multiple purposes in the validation context: ensuring comprehensive test coverage, supporting impact assessment for changes, and providing evidence of validation completeness. Section 6 positions traceability as fundamental validation infrastructure rather than optional documentation enhancement.
For organizations accustomed to simplified validation approaches where test cases are developed independently of detailed requirements, this traceability requirement represents a significant process change requiring tool investment and training.
6.6: Configuration—Separating Standard from Custom
The final subsection addresses configuration management by requiring clear documentation of “what functionality, if any, is modified or added by configuration of a system.” This requirement recognizes that most modern pharmaceutical systems involve significant configuration rather than custom development, and that configuration decisions have direct impact on validation scope and approaches.
The distinction between standard system functionality and configured functionality is crucial for validation planning. Standard functionality may be covered by vendor testing and certification, while configured functionality requires user validation. Section 6 requires this distinction to be explicit and documented.
The requirement for “controlled configuration specification” separate from requirements documentation reflects recognition that configuration details require different management approaches than functional requirements. Configuration specifications must reflect the actual system implementation rather than desired capabilities.
Comparison with GAMP 5: Evolution Becomes Revolution
Philosophical Alignment with Practical Divergence
Section 6 maintains GAMP 5’s fundamental philosophy—risk-based validation supported by comprehensive requirements documentation—while dramatically changing implementation expectations. Both frameworks emphasize user ownership of requirements, lifecycle management, and traceability as essential validation elements. However, the regulatory context of Annex 11 transforms voluntary guidance into enforceable obligation.
GAMP 5’s flexibility in requirements categorization and documentation approaches reflects its role as guidance suitable for diverse organizational contexts and system types. Section 6’s prescriptive approach reflects regulatory recognition that flexibility has often been interpreted as optionality, leading to inadequate requirements documentation that fails to support effective validation.
The risk-based approach remains central to both frameworks, but Section 6 establishes minimum standards that apply regardless of risk assessment outcomes. While GAMP 5 might suggest that low-risk systems require minimal requirements documentation, Section 6 mandates coverage of nine requirement areas for all GMP systems.
Documentation Structure and Content
GAMP 5’s traditional document hierarchy—URS, Functional Specification, Design Specification—becomes more fluid under Section 6, which focuses on ensuring comprehensive coverage rather than prescribing specific document structures. This reflects recognition that modern development approaches, including agile and DevOps practices, may not align with traditional waterfall documentation models.
However, Section 6’s explicit enumeration of requirement types provides more prescriptive guidance than GAMP 5’s flexible approach. Where GAMP 5 might allow organizations to define requirement categories based on system characteristics, Section 6 mandates coverage of operational, functional, data integrity, technical, interface, performance, availability, security, and regulatory requirements.
The emphasis on process maps, data flow diagrams, and use cases reflects modern system complexity where understanding interactions and dependencies is essential for effective validation. GAMP 5 recommends these approaches for complex systems; Section 6 suggests their use “where relevant” for all systems.
Vendor and Service Provider Management
Both frameworks emphasize user responsibility for requirements even when vendors provide initial documentation. However, Section 6 uses stronger language about user ownership and control, reflecting increased regulatory concern about organizations that delegate requirements definition to vendors without adequate oversight.
GAMP 5’s guidance on supplier assessment and leveraging vendor documentation remains relevant under Section 6, but the regulatory requirement for user ownership and approval creates higher barriers for simply accepting vendor-provided documentation as sufficient.
Implementation Challenges for CSV Professionals
Organizational Capability Development
Most pharmaceutical organizations will require significant capability development to meet Section 6 requirements effectively. Traditional validation teams focused on testing and documentation must develop requirements engineering capabilities comparable to those found in software development organizations.
This transformation requires investment in requirements management tools, training for validation professionals, and establishment of requirements governance processes. Organizations must develop capabilities for requirements elicitation, analysis, specification, validation, and change management throughout the system lifecycle.
The traceability requirement particularly challenges organizations accustomed to informal relationships between requirements and test cases. Automated traceability management requires tool investments and process changes that many validation teams are unprepared to implement.
Integration with Existing Validation Approaches
Section 6 requirements must be integrated with existing validation methodologies and documentation structures. Organizations following traditional IQ/OQ/PQ approaches must ensure that requirements documentation supports and guides qualification activities rather than existing as parallel documentation.
The requirement for requirements to “form the very basis for qualification and validation” means that test cases must be explicitly derived from and traceable to documented requirements. This may require significant changes to existing qualification protocols and test scripts.
Organizations using risk-based validation approaches aligned with GAMP 5 guidance will find philosophical alignment with Section 6 but must adapt to more prescriptive requirements for documentation content and structure.
Technology and Tool Requirements
Effective implementation of Section 6 requirements typically requires requirements management tools capable of supporting specification, traceability, change control, and lifecycle management. Many pharmaceutical validation teams currently lack access to such tools or experience in their use.
Tool selection must consider integration with existing validation platforms, support for regulated environments, and capabilities for automated traceability maintenance. Organizations may need to invest in new validation platforms or significantly upgrade existing capabilities.
The emphasis on maintaining requirements throughout the system lifecycle requires tools that support ongoing requirements management rather than just initial documentation. This may conflict with validation approaches that treat requirements as static inputs to qualification activities.
Strategic Implications for the Industry
Convergence of Software Engineering and Pharmaceutical Validation
Section 6 represents convergence between pharmaceutical validation practices and mainstream software engineering approaches. Requirements engineering, long established in software development, becomes mandatory for pharmaceutical computerized systems regardless of development approach or vendor involvement.
This convergence benefits the industry by leveraging proven practices from software engineering while maintaining the rigor and documentation requirements essential for regulated environments. However, it requires pharmaceutical organizations to develop capabilities traditionally associated with software development rather than manufacturing and quality assurance.
The result should be more robust validation practices better aligned with modern system development approaches and capable of supporting the complex, interconnected systems that characterize contemporary pharmaceutical operations.
Vendor Relationship Evolution
Section 6 requirements will reshape relationships between pharmaceutical companies and system vendors. The requirement for user ownership of requirements documentation means that vendors must support more sophisticated requirements management processes rather than simply providing generic specifications.
Vendors that can demonstrate alignment with Section 6 requirements through comprehensive documentation, traceability tools, and support for user customization will gain competitive advantages. Those that resist pharmaceutical-specific requirements management approaches may find their market opportunities limited.
The emphasis on configuration management will drive vendors to provide clearer distinctions between standard functionality and customer-specific configurations, supporting more effective validation planning and execution.
The Regulatory Codification of Modern Validation
Section 6 of the draft Annex 11 represents the regulatory codification of modern computerized system validation practices. What GAMP 5 recommended through guidance, Annex 11 mandates through regulation. What was optional becomes obligatory; what was flexible becomes prescriptive; what was best practice becomes compliance requirement.
For CSV professionals, Section 6 provides regulatory support for comprehensive validation approaches while raising the stakes for inadequate implementation. Organizations that have struggled to implement effective requirements management now face regulatory obligation rather than just professional guidance.
The transformation from guidance to regulation eliminates organizational discretion about requirements documentation quality and comprehensiveness. While risk-based approaches remain valid for scaling validation effort, minimum standards now apply regardless of risk assessment outcomes.
Success under Section 6 requires pharmaceutical organizations to embrace software engineering practices for requirements management while maintaining the documentation rigor and process control essential for regulated environments. This convergence benefits the industry by improving validation effectiveness while ensuring compliance with evolving regulatory expectations.
The industry faces a choice: proactively develop capabilities to meet Section 6 requirements or reactively respond to inspection findings and enforcement actions. For organizations serious about digital transformation and validation excellence, Section 6 provides a roadmap for regulatory-compliant modernization of validation practices.
Requirement Area
Draft Annex 11 Section 6
GAMP 5 Requirements
Key Implementation Considerations
System Requirements Documentation
Mandatory – Must establish and approve system requirements (URS)
Recommended – URS should be developed based on system category and complexity
Organizations must document requirements for ALL GMP systems, regardless of size or complexity
Risk-Based Approach
Extent and detail must be commensurate with risk, complexity, and novelty
Risk-based approach fundamental – validation effort scaled to risk
Risk assessment determines documentation detail but cannot eliminate requirement categories
Functional Requirements
Must include 9 specific requirement types: operational, functional, data integrity, technical, interface, performance, availability, security, regulatory
Functional requirements should be SMART (Specific, Measurable, Achievable, Realistic, Testable)
All 9 areas must be addressed; risk determines depth, not coverage
Traceability Requirements
Documented traceability between requirements, design specs, and test cases required
Traceability matrix recommended – requirements linked through design to testing
Requires investment in traceability tools and processes for complex systems
Requirement Ownership
Regulated user must take ownership even if vendor provides initial requirements
User ownership emphasized, even for purchased systems
Cannot simply accept vendor documentation; must customize and formally approve
Lifecycle Management
Requirements must be updated and maintained throughout system lifecycle
Requirements managed through change control throughout lifecycle
Requires ongoing requirements management process, not just initial documentation
Configuration Management
Configuration options must be described in requirements; chosen configuration documented in controlled spec
Configuration specifications separate from URS
Must clearly distinguish between standard functionality and configured features
Vendor-Supplied Requirements
Vendor requirements must be reviewed, approved, and owned by regulated user
Supplier assessment required – leverage supplier documentation where appropriate
Higher burden on users to customize vendor documentation for specific GMP needs
Validation Basis
Updated requirements must form basis for system qualification and validation
Requirements drive validation strategy and testing scope
Requirements become definitive specification against which system performance is measured
Ready or not, the EU’s draft revision of Annex 11 is moving toward finalization, and its brand-new Section 13 on electronic signatures is a wake-up call for anyone still treating digital authentication as just Part 11 with an accent. In this post I will take a deep dive into what’s changing, why it matters, and how to keep your quality system out of the regulatory splash zone.
Section 13 turns electronic signatures from a check-the-box formality into a risk-based, security-anchored discipline. Think multi-factor authentication, time-zone stamps, hybrid wet-ink safeguards, and explicit “non-repudiation” language—all enforced at the same rigor as system login. If your current SOPs still assume username + password = done, it’s time to start planning some improvements.
Why the Rewrite?
Tech has moved on: Biometric ID, cloud PaaS, and federated identity management were sci-fi when the 2011 Annex 11 dropped.
Threat landscape: Ransomware and credential stuffing didn’t exist at today’s scale. Regulators finally noticed.
Global convergence: The FDA’s Computer Software Assurance (CSA) draft and PIC/S data-integrity guides pushed the EU to level up.
Must equal or exceed login strength; first sign = full re-auth, subsequent signs = pwd/biometric; smart-card-only = banned
Two identification components
Forces MFA or biometrics; goodbye “remember me” shortcuts
Time & Time-Zone
Date + time (manual OK)
Auto-captured and time-zone logged
Date + time (no TZ)
Multisite ops finally get defensible chronology
Signature Meaning Prompt
Not required
System must ask user for purpose (approve, review…)
Required but less prescriptive
Eliminates “mystery clicks” that auditors love to exploit
Manifestation Elements
Minimal
Full name, username, role, meaning, date/time/TZ
Name, date, meaning
Closes attribution gaps; boosts ALCOA+ “Legible”
Indisputability Clause
“Same impact”
Explicit non-repudiation mandate
Equivalent legal weight
Sets the stage for eIDAS/federated ID harmonization
Record Linking After Change
Permanent link
If record altered post-sign, signature becomes void/flagged
Link cannot be excised
Ends stealth edits after approval
Hybrid Wet-Ink Control
Silent
Hash code or similar to break link if record changes
Silent
Lets you keep occasional paper without tanking data integrity
Open Systems / Trusted Services
Silent
Must comply with “national/international trusted services” (read: eIDAS)
Extra controls, but legacy wording
Validates cloud signing platforms out of the box
The Implications
Multi-Factor Authentication (MFA) Is Now Table Stakes
Because the draft explicitly bars any authentication method that relies solely on a smart card or a static PIN, every electronic signature now has to be confirmed with an additional, independent factor—such as a password, biometric scan, or time-limited one-time code—so that the credential used to apply the signature is demonstrably different from the one that granted the user access to the system in the first place.
Time-Zone Logging Kills Spreadsheet Workarounds
One of the more subtle but critical updates in Draft Annex 11’s Section 13.4 is the explicit requirement for automatic logging of the time zone when electronic signatures are applied. Unlike previous guidance—whether under the 2011 Annex 11 or 21 CFR Part 11—that only mandated the capture of date and time (often allowing manual entry or local system time), the draft stipulates that systems must automatically capture the precise time and associated time zone for each signature event. This seemingly small detail has monumental implications for data integrity, traceability, and regulatory compliance. Why does this matter? For global pharmaceutical operations spanning multiple time zones, manual or local-only timestamps often create ambiguous or conflicting audit trails, leading to discrepancies in event sequencing. Companies relying on spreadsheets or legacy systems that do not incorporate time zone information effectively invite errors where a signature in one location appears to precede an earlier event simply due to zone differences. This ambiguity can undermine the “Contemporaneous” and “Enduring” principles of ALCOA+, principles the draft Annex 11 explicitly reinforces throughout electronic signature requirements. By mandating automated, time zone-aware timestamping, Draft Annex 11 Section 13.4 ensures that electronic signature records maintain a defensible and standardized chronology across geographies, eliminating the need for cumbersome manual reconciliation or retrospective spreadsheet corrections. This move not only tightens compliance but also supports modern, centralized data review and analytics where uniform timestamping is essential. If your current systems or SOPs rely on manual date/time entry or overlook time zone logging, prepare for significant system and procedural updates to meet this enhanced expectation once the draft Annex 11 is finalized. .
Hybrid Records Are Finally Codified
If you still print a batch record for wet-ink QA approval, Section 13.9 lets you keep the ritual—but only if a cryptographic hash or similar breaks when someone tweaks the underlying PDF. Expect a flurry of DocuSign-scanner-hash utilities.
Open-System Signatures Shift Liability
Draft Annex 11’s Section 13.2 represents perhaps the most strategically significant change in electronic signature liability allocation since 21 CFR Part 11 was published in 1997. The provision states that “Where the system owner does not have full control of system accesses (open systems), or where required by other legislation, electronic signatures should, in addition, meet applicable national and international requirements, such as trusted services”. This seemingly simple sentence fundamentally reshapes liability relationships in modern pharmaceutical IT architectures.
Defining the Open System Boundary
The draft Annex 11 adopts the 21 CFR Part 11 definition of open systems—environments where system owners lack complete control over access and extends it into contemporary cloud, SaaS, and federated identity scenarios. Unlike the original Part 11 approach, which merely required “additional measures such as document encryption and use of appropriate digital signature standards”, Section 13.2 creates a positive compliance obligation by mandating adherence to “trusted services” frameworks.
This distinction is critical: while Part 11 treats open systems as inherently risky environments requiring additional controls, draft Annex 11 legitimizes open systems provided they integrate with qualified trust service providers. Organizations no longer need to avoid cloud-based signature services; instead, they must ensure those services meet eIDAS-qualified standards or equivalent national frameworks.
The Trusted Services Liability Transfer
Section 13.2’s reference to “trusted services” directly incorporates European eIDAS Regulation 910/2014 into pharmaceutical GMP compliance, creating what amounts to a liability transfer mechanism. Under eIDAS, Qualified Trust Service Providers (QTSPs) undergo rigorous third-party audits, maintain certified infrastructure, and provide legal guarantees about signature validity and non-repudiation. When pharmaceutical companies use eIDAS-qualified signature services, they effectively transfer signature validity liability from their internal systems to certified external providers.
This represents a fundamental shift from the 21 CFR Part 11 closed-system preference, where organizations maintained complete control over signature infrastructure but also bore complete liability for signature failures. Draft Annex 11 acknowledges that modern pharmaceutical operations often depend on cloud service providers, federated authentication systems, and external trust services—and provides a regulatory pathway to leverage these technologies while managing liability exposure.
Practical Implications for SaaS Platforms
The most immediate impact affects organizations using Software-as-a-Service platforms for clinical data management, quality management, or document management. Under current Annex 11 and Part 11, these systems often require complex validation exercises to demonstrate signature integrity, with pharmaceutical companies bearing full responsibility for signature validity even when using external platforms.
Section 13.2 changes this dynamic by validating reliance on qualified trust services. Organizations using platforms like DocuSign, Adobe Sign, or specialized pharmaceutical SaaS providers can now satisfy Annex 11 requirements by ensuring their chosen platforms integrate with eIDAS-qualified signature services. The pharmaceutical company’s validation responsibility shifts from proving signature technology integrity to verifying trust service provider qualifications and proper integration.
Integration with Identity and Access Management
Draft Annex 11’s Section 11 (Identity and Access Management) works in conjunction with Section 13.2 to support federated identity scenarios common in modern pharmaceutical operations. Organizations can now implement single sign-on (SSO) systems with external identity providers, provided the signature components integrate with trusted services. This enables scenarios where employees authenticate through corporate Active Directory systems but execute legally binding signatures through eIDAS-qualified providers.
The liability implications are significant: authentication failures become the responsibility of the identity provider (within contractual limits), while signature validity becomes the responsibility of the qualified trust service provider. The pharmaceutical company retains responsibility for proper system integration and user access controls, but shares technical implementation liability with certified external providers.
Cloud Service Provider Risk Allocation
For organizations using cloud-based LIMS, MES, or quality management systems, Section 13.2 provides regulatory authorization to implement signature services hosted entirely by external providers. Cloud service providers offering eIDAS-compliant signature services can contractually accept liability for signature technical implementation, cryptographic integrity, and legal validity—provided they maintain proper trust service qualifications.
This risk allocation addresses a long-standing concern in pharmaceutical cloud adoption: the challenge of validating signature infrastructure owned and operated by external parties. Under Section 13.2, organizations can rely on qualified trust service provider certifications rather than conducting detailed technical validation of cloud provider signature implementations.
Harmonization with Global Standards
Section 13.2’s “national and international requirements” language extends beyond eIDAS to encompass other qualified electronic signature frameworks. This includes Swiss ZertES standards and Canadian digital signature regulations,. Organizations operating globally can implement unified signature platforms that satisfy multiple regulatory requirements through single trusted service provider integrations.
The practical effect is regulatory arbitrage: organizations can choose signature service providers based on the most favorable combination of technical capabilities, cost, and regulatory coverage, rather than being constrained by local regulatory limitations.
Supplier Assessment Transformation
Draft Annex 11’s Section 7 (Supplier and Service Management) requires comprehensive supplier assessment for computerized systems. However, Section 13.2 creates a qualified exception for eIDAS-certified trust service providers: organizations can rely on third-party certification rather than conducting independent technical assessments of signature infrastructure.
This significantly reduces supplier assessment burden for signature services. Instead of auditing cryptographic implementations, hardware security modules, and signature validation algorithms, organizations can verify trust service provider certifications and assess integration quality. The result: faster implementation cycles and reduced validation costs for signature-enabled systems.
Audit Trail Integration Considerations
The liability shift enabled by Section 13.2 affects audit trail management requirements detailed in draft Annex 11’s expanded Section 12 (Audit Trails). When signature events are managed by external trust service providers, organizations must ensure signature-related audit events are properly integrated with internal audit trail systems while maintaining clear accountability boundaries.
Qualified trust service providers typically provide comprehensive signature audit logs, but organizations remain responsible for correlation with business process audit trails. This creates shared audit trail management where signature technical events are managed externally but business context remains internal responsibility.
Competitive Advantages of Early Adoption
Organizations that proactively implement Section 13.2 requirements gain several strategic advantages:
Reduced Infrastructure Costs: Elimination of internal signature infrastructure maintenance and validation overhead
Enhanced Security: Leverage specialized trust service provider security expertise and certified infrastructure
Global Scalability: Unified signature platforms supporting multiple regulatory jurisdictions through single provider relationships
Accelerated Digital Transformation: Faster deployment of signature-enabled processes through validated external services
Risk Transfer: Contractual liability allocation with qualified external providers rather than complete internal risk retention
Section 13.2 transforms open system electronic signatures from compliance challenges into strategic enablers of digital pharmaceutical operations. By legitimizing reliance on qualified trust services, the draft Annex 11 enables organizations to leverage best-in-class signature technologies while managing regulatory compliance and liability exposure through proven external partnerships. The result: more secure, cost-effective, and globally scalable electronic signature implementations that support advanced digital quality management systems.
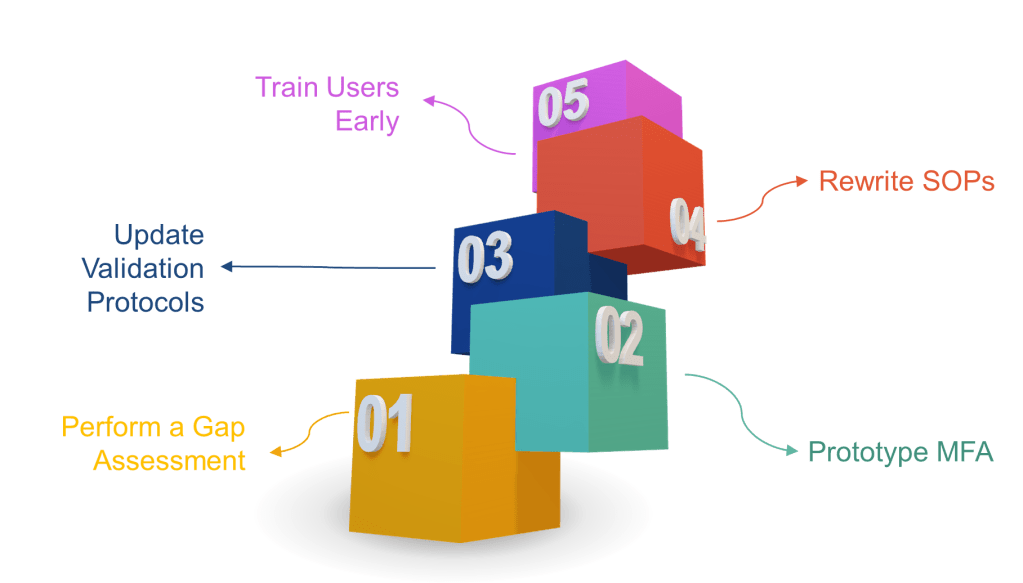
How to Get Ahead (Instead of Playing Cleanup)
Perform a gap assessment now—map every signature point to the new rules.
Prototype MFA in your eDMS or MES. If users scream about friction, remind them that ransomware is worse.
Update validation protocols to include time-zone, hybrid record, and non-repudiation tests.
Rewrite SOPs to include signature-meaning prompts and periodic access-right recertification.
Train users early. A 30-second “why you must re-authenticate” explainer video beats 300 deviations later.
Final Thoughts
The draft Annex 11 doesn’t just tweak wording—it yanks electronic signatures into the 2020s. Treat Section 13 as both a compliance obligation and an opportunity to slash latent data-integrity risk. Those who adapt now will cruise through 2026/2027 inspections while the laggards scramble for remediation budgets.
I think folks tend to fall into a trap when it comes to equipment and GAMP5, automatically assuming that because it is equipment it must be Category 3. Oh, how that can lead to problems.
When thinking about equipment it is best to think in terms of “No Configuration” and ” Low Configuration” software. This terminology is used to describe software that requires little to no configuration or customization to meet the user’s needs.
No Configuration(NoCo) aligns with GAMP 5 Category 3 software, which is described as “Non-Configured Products”. These are commercial off-the-shelf software applications that are used as-is, without any customization or with only minimal parameter settings. My microwave is NoCo.
Low Configuration(LoCo) typically falls between Category 3 and Category 4 software. It refers to software that requires some configuration, but not to the extent of fully configurable systems. My PlayStation is LoCo.
The distinction between these categories is important for determining the appropriate validation approach:
Category 3 (NoCo) software generally requires less extensive validation efforts, as it is used without significant modifications. Truly it can be implicit testing.
Software with low configuration may require a bit more scrutiny in validation, but still less than fully configurable or custom-developed systems.
Remember that GAMP 5 emphasizes a continuum approach rather than strict categorization. The level of validation effort should be based on the system’s impact on patient safety, product quality, and data integrity, as well as the extent of configuration or customization.
When is Something Low Configuration?
Low Configuration refers to software that requires minimal setup or customization to meet user needs, falling between Category 3 (Non-Configured Products) and Category 4 (Configured Products) software. Here’s a breakdown of what counts as low configuration:
Parameter settings: Software that allows basic parameter adjustments without altering core functionality.
Limited customization: Applications that permit some tailoring to specific workflows, but not extensive modifications.
Standard modules: Software that uses pre-built, configurable modules to adapt to business processes.
Default configurations: Systems that can be used with supplier-provided default settings or with minor adjustments.
Simple data input: Applications that allow input of specific data or ranges, such as electronic chart recorders with input ranges and alarm setpoints.
Basic user interface customization: Software that allows minor changes to the user interface without altering underlying functionality.
Report customization: Systems that permit basic report formatting or selection of data fields to display.
Simple workflow adjustments: Applications that allow minor changes to predefined workflows without complex programming.
It’s important to note that the distinction between low configuration and more extensive configuration (Category 4) can sometimes be subjective. The key is to assess the extent of configuration required and its impact on the system’s core functionality and GxP compliance. Organizations should document their rationale for categorization in system risk assessments or validation plans.
Attribute
Category 3 (No Configuration)
Low Configuration
Category 4
Configuration Level
No configuration
Minimal configuration
Extensive configuration
Parameter Settings
Fixed or minimal
Basic adjustments
Complex adjustments
Customization
None
Limited
Extensive
Modules
Pre-built, non-configurable
Standard, slightly configurable
Highly configurable
Default Settings
Used as-is
Minor adjustments
Significant modifications
Data Input
Fixed format
Simple data/range input
Complex data structures
User Interface
Fixed
Basic customization
Extensive customization
Workflow Adjustments
None
Minor changes
Significant alterations
User Account Management
Basic, often single-user
Limited user roles and permissions
Advanced user management with multiple roles and access levels
Report Customization
Pre-defined reports
Basic formatting/field selection
Advanced report design
Example Equipment
pH meter
Electronic chart recorder
Chromatography data system
Validation Effort
Minimal
Moderate
Extensive
Risk Level
Low
Low to Medium
Medium to High
Supplier Documentation
Heavily relied upon
Partially relied upon
Supplemented with in-house testing
Here’s the thing to be aware of, a lot of equipment these days is more category 4 than 3, as the manufacturers include all sorts of features, such as user account management and trending and configurable reports. And to be frank, I’ve seen too many situations where Programmable Logic Controllers (PLCs) didn’t take into account all that configuration from standard function libraries to control specific manufacturing processes.
Your methodology needs to keep up with the technological growth curve.