On January 19, 2026, the EMA GMP/GDP Inspectors Working Group and PIC/S published a concept paper proposing a targeted revision of EU GMP Annex 15—Qualification and Validation. The public consultation opened on February 9 and runs through April 9, 2026. If you work in active substance manufacturing, or if your drug product quality depends on active substance quality—which is to say, if you work in this industry at all—this document deserves your attention.
The headline is straightforward: Annex 15 will become mandatory for active substance manufacturers. But what makes this revision significant isn’t just the shift from optional to mandatory. It’s what the shift reveals about where the regulatory landscape is heading, and how many of the themes I’ve been writing about on this blog—living risk management, control strategy as connective tissue, the validation lifecycle as a knowledge system—are now being codified into explicit regulatory expectations for a sector that has, frankly, lagged behind.
The Nitrosamine Wake-Up Call
The revision traces its origin directly to the N-nitrosamine crisis in sartan medicines. The EMA’s June 2020 lessons-learnt report was unsparing: one root cause of nitrosamine contamination was “the lack of sufficient process and product knowledge during the development stage and GMP deficiencies by active substance manufacturers, including inadequate investigation of quality issues and insufficient contamination control measures”. This wasn’t a novel finding at the time, but the sartans case gave regulators the political and scientific impetus to act.
Paragraph 4.2.2 of that lessons-learnt report specifically recommended making Annex 15 mandatory for active substance manufacturers to address the shortcomings identified during inspections. It took several years of deliberation—the GMP/GDP IWG formally agreed to proceed at its 115th meeting in September 2024—but the wheels are now turning.
The lesson here is one I’ve returned to repeatedly: knowledge gaps don’t stay dormant. They surface as deviations, contamination events, and regulatory actions. The sartans crisis was, at its core, a failure of process understanding and control strategy—areas where Annex 15 is now being strengthened precisely because too many active substance manufacturers treated validation as peripheral rather than foundational.
What the Concept Paper Actually Proposes
Let me walk through the key elements of the proposed revision, because the specifics matter more than the headline.
Scope Extension
The revised Annex 15 will apply to manufacturers of both chemical and biological active substances. EU and PIC/S inspectorates will enforce compliance during regulatory inspections. This is a paradigm shift for API manufacturers who have historically operated under Part II of the EU GMP Guide with Annex 15 as optional supplementary guidance. The concept paper is clear: “Although annex 15 is not currently mandatory for AS manufacturers, the applicability of its principles in this sector is generally recognised”. In other words, the expectation already existed—now it will have enforcement teeth.
Validation Master File, Policy, and Change Control
The concept paper proposes extending the Validation Master File, the Qualification and Validation Policy, and formal change control requirements to active substance manufacturers. These aren’t new concepts for drug product manufacturers, but their extension to AS manufacturers signals a regulatory expectation of structured, documented validation programs rather than ad hoc approaches.
Change control, in particular, is described as “an important part of knowledge management”. This language is deliberate and echoes what I’ve been writing about in the context of control strategies and the feedback-feedforward controls hub: change control isn’t bureaucratic overhead—it’s the mechanism through which accumulated process knowledge is preserved, evaluated, and applied.
Validation Discrepancies
The revision will extend the requirement to investigate results that fail to meet pre-defined acceptance criteria during validation activities. This extension, the concept paper notes, “will promote AS manufacturers to have a more in-depth knowledge of their processes.” This is one of the most quietly important provisions. In my experience, the gap between drug product and active substance manufacturers is often widest in investigation rigor. Robust investigation of validation failures isn’t just about compliance—it’s about generating the process knowledge that underpins meaningful control strategies.
Qualification Stages: URS, FAT/SAT, DQ/IQ/OQ/PQ
The concept paper extends the formal qualification lifecycle—User Requirements Specifications, Factory Acceptance Testing, Site Acceptance Testing, and the traditional DQ/IQ/OQ/PQ sequence—to active substance manufacturing. For those of us who have worked in the ASTM E2500 and ISPE commissioning and qualification frameworks, this is a natural evolution. As I discussed in my posts on CQV and engineering runs, these qualification stages aren’t separate activities—they form a continuum where each stage builds on the knowledge generated in the previous one. Extending this structured approach to API manufacturing strengthens the design-validation continuum that is essential for robust control strategies.
Process Validation: Development, Concurrent Validation, CPV, and Recovery
Several process validation enhancements are proposed:
Emphasis on robust process development: Clarifying that validation begins with development, not with the first PPQ batch.
Clarification of concurrent validation: Tightening expectations on when and how concurrent validation may be used.
Continuous process verification and hybrid approaches: Extending Stage 3/CPV thinking to active substance manufacturing.
Recovery of materials and solvents: Extending validation requirements to solvent and material recovery processes.
Supplier qualification: Emphasizing the role of supplier qualification in the validation ecosystem.
Periodic review: Reinforcing the expectation that validation is a lifecycle activity, not a one-time event.
This aligns directly with what I wrote about in Continuous Process Verification (CPV) Methodology and Tool Selection: CPV is “not an isolated activity but a continuation of the knowledge gained in earlier stages”. The lifecycle approach—Process Design (Stage 1), Process Qualification (Stage 2), Continued Process Verification (Stage 3)—is being explicitly extended to a sector that has too often treated validation as a discrete project rather than an ongoing program.
Transport Verification
The revision extends expectations for transport verification, linking GMP with Good Distribution Practices (GDP) for active substances. This addresses a gap that has been hiding in plain sight: product knowledge must include understanding of how transportation affects quality. For biologically-derived active substances in particular, this provision acknowledges that the supply chain is part of the process, not external to it.
ICH Q9 (R1) Integration
The concept paper mandates incorporation of ICH Q9 (R1) quality risk management principles throughout validation and qualification activities. Specifically:
QRM in the design and validation/qualification of monitoring systems
Risk review activities to support ongoing validation and qualification
Emphasis on QRM in the context of traditional processes
This integration is overdue. As I discussed in Living Risk in the Validation Lifecycle and Risk Management is a Living Process, effective risk management isn’t a one-time exercise performed during design—it’s a living system that evolves throughout the product lifecycle. ICH Q9 (R1) itself emphasizes that “the level of effort, formality and documentation of the quality risk management process should be commensurate with the level of risk.” It introduces the importance-complexity-uncertainty framework for calibrating risk assessment rigor. The Annex 15 revision will make these principles explicitly applicable to qualification and validation decisions in active substance manufacturing.
Why This Matters: The Industry-Wide Implications
Closing the Knowledge Gap
The fundamental driver of this revision is a knowledge deficit. The nitrosamine crisis exposed what many of us already suspected: a significant number of active substance manufacturers lacked the process understanding necessary to predict, prevent, and detect quality problems. Making Annex 15 mandatory doesn’t automatically create knowledge, but it creates the structural requirements—validation master plans, formal qualification stages, investigation requirements, CPV programs—that force organizations to build and maintain it.
As I explored in Control Strategies, control strategies represent “the central mechanism through which pharmaceutical companies ensure quality, manage risk, and leverage knowledge”. Without the foundational process knowledge that structured validation generates, control strategies are hollow documents. The Annex 15 revision, by mandating the validation activities that generate this knowledge for active substance manufacturers, strengthens the entire control strategy ecosystem from the ground up.
From Compliance Burden to Audit Readiness
In my analysis of the 2025 State of Validation data, I noted a striking reversal: audit readiness has overtaken compliance burden as the industry’s primary validation challenge. This shift reflects a maturation of validation programs—organizations are moving from the scramble to implement validation to the discipline of sustaining it. The Annex 15 revision will push active substance manufacturers through a similar maturation arc. The initial impact will feel like compliance burden. But the long-term trajectory, if organizations approach it with the right mindset, is toward sustained audit readiness grounded in genuine process knowledge.
Risk Management as the Connective Thread
The integration of ICH Q9 (R1) throughout the revised Annex 15 reinforces a theme I’ve been tracking across multiple regulatory developments: risk management is no longer a supporting tool—it’s the connective thread that runs through every quality decision. The parallel revision of EudraLex Chapter 1, the new Annex 11 requirements for computerized systems, and the forthcoming Annex 22 for artificial intelligence all place quality risk management at their center. The Annex 15 revision ensures that qualification and validation are no exception.
This convergence means that organizations need integrated risk management capabilities—not siloed risk assessments performed by different teams for different purposes, but a coherent QRM framework that connects design risk, process risk, facility risk, and supply chain risk into a unified picture. As I wrote in my piece on risk management and change management: “Risk management leads to change management. Change management contains risk management”. The revised Annex 15 makes this cycle explicit for active substance manufacturers.
The Control Strategy Connection
Perhaps the most significant implication is how this revision strengthens the link between validation and control strategy. In Control Strategies, I described how control strategies occupy “that critical program-level space between overarching quality policies and detailed operational procedures” and serve as “the blueprint for how quality will be achieved, maintained, and improved throughout a product’s lifecycle”.
The Annex 15 revision reinforces every dimension of this blueprint for active substance manufacturing:
Validation Master File → documents the overall validation approach and connects it to the control strategy
Formal qualification stages → ensure that facility and equipment design supports the intended control strategy
Process validation with CPV → generates the ongoing data that validates and refines the control strategy
Investigation of failures → feeds new knowledge back into the control strategy through the feedback loop
Change control as knowledge management → ensures that the control strategy evolves based on accumulated understanding
Transport verification → extends the control strategy to encompass the supply chain
This is the feedback-feedforward controls hub in action. Each element of the revised Annex 15 either generates knowledge that feeds into the control strategy or applies knowledge from the control strategy to operational decisions.
The PLCM Document and Established Conditions
Looking forward, this revision also has implications for how active substance manufacturers engage with ICH Q12 concepts. As I discussed in my recent post on the Product Lifecycle Management (PLCM) document, the distinction between comprehensive control strategy elements and Established Conditions is critical for enabling continuous improvement. Active substance manufacturers who build robust validation and knowledge management programs now—in response to the Annex 15 revision—will be better positioned to participate in lifecycle management frameworks that reward process understanding with regulatory flexibility.
The concept paper’s emphasis on “change control as an important part of knowledge management” directly supports this trajectory. Organizations that treat change control as a bureaucratic hurdle will miss the point. Those that treat it as a knowledge capture mechanism will find themselves building the foundation for more sophisticated lifecycle management.
The Timeline and What to Do Now
The proposed timetable is aggressive:
Milestone
Date
Concept paper public consultation
February – April 2026
Draft guideline consultation
April – June 2026
EMA GMP/GDP IWG endorsement
July 2026
Publication by European Commission
December 2026
PIC/S adoption
December 2026
The concept paper includes four stakeholder questions that are worth engaging with seriously:
What is the current level of use of Annex 15 principles in active substance manufacturing?
What would be the impact of making Annex 15 mandatory?
What is the current understanding and use of ICH Q9 (R1) in active substance manufacturing?
What would be the impact of incorporating Q9 (R1)?
If you manufacture active substances—or if you’re a drug product manufacturer who depends on active substance suppliers—now is the time to:
Perform a gap assessment against the current Annex 15 requirements, assuming mandatory application
Evaluate your Validation Master Plan or equivalent program documentation for active substance operations
Review your qualification lifecycle to ensure URS, FAT/SAT, and formal qualification stages are documented and traceable
Assess your CPV program for active substance processes—does it exist? Is it generating actionable knowledge?
Examine your investigation process for validation failures against pre-defined acceptance criteria
Review your QRM integration into qualification and validation activities against ICH Q9 (R1) expectations
Engage with the public consultation by the April 9, 2026 deadline
The Bigger Picture
The concept paper notes that the GMP/GDP IWG also agreed that “a comprehensive review of Annex 15 should be initiated in the future, once the current targeted revision is finished”. This targeted revision is just the beginning. A full-scope revision will likely address the broader evolution of validation thinking—digital systems, advanced analytics, platform approaches—that I’ve been tracking in posts on the evolving validation landscape.
The world of validation is no longer controlled by periodic updates or leisurely transitions. Change is the new baseline. The Annex 15 revision is another data point in a pattern that includes the Annex 1 overhaul, the Annex 11 modernization, the introduction of Annex 22, the ICH Q9 (R1) revision, and the convergence of global regulators around lifecycle, risk-based, and knowledge-driven approaches to quality.
For active substance manufacturers, the message is clear: the era of treating validation as optional supplementary guidance is over. For the rest of us, the message is equally important: the quality of our medicines depends on the quality of knowledge throughout the supply chain, and regulators are now ensuring that the structural requirements to generate and maintain that knowledge extend to every link in the chain.
The draft revision of Eudralex Volume 4 Chapter 1 marks a substantial evolution from the current version, reflecting regulatory alignment with ICH Q9(R1), enhanced risk-based approaches, and a new emphasis on knowledge management, proactive risk detection, and supply chain resilience.
Core Differences at a Glance
The draft update integrates advances in global quality science—especially from ICH Q9(R1)—anchoring the Pharmaceutical Quality System (PQS) more firmly in knowledge management and risk management practice.
Proactive risk identification and mitigation are highlighted, reflecting the need to anticipate supply disruptions and quality failures, beyond routine compliance.
The requirements for Product Quality Review (PQR) are clarified, notably in how to handle grouped products and limited-batch scenarios, enhancing operational clarity for diverse manufacturing models.
Philosophical Shift: From Compliance to Dynamic Risk Management
Where the current Chapter 1 (in force since 2013) framed the PQS largely as a static structure of roles, documentation, and reviews, the draft version pivots toward a learning organization approach: knowledge acquisition, use, and feedback become core system elements.
Emphasis is now placed on systematic knowledge management as both a regulatory and operational priority. This serves as an overt marker of quality system maturity, intended to reduce “invisible failures” and foster analytical vigilance—aligning closely with falsifiable quality frameworks.
Risk-Based Decision-Making: Explicit and Actionable
The revision operationalizes risk-based thinking by mandating scientific rationale for risk decisions and clarifying expectations for proportionality in risk assessment. The regulator’s intent is clear: risk management can no longer be a box-checking exercise, but must be demonstrably linked to daily site operations and lifecycle decisions.
This brings the PQS into closer alignment with both the adaptive toolbox and the take-the-best heuristics: decisive focus on the most causally relevant risk vectors rather than exhaustive factor listing, echoing playbooks for effective investigation and CAPA prioritization.
Product Quality Review (PQR) and Batch Grouping
Clarification is provided in the revised text on how to perform quality reviews for products manufactured in small numbers or as grouped products, a challenge long met with uncertainty. The draft provides operational guidance, aiming to resolve ambiguities around the statistical and process review requirements for product families and low-volume production.
Supply Chain Resilience, Shortage Prevention, and Knowledge Networks
The draft gives unprecedented attention to shortage prevention and supply chain risk. Manufacturers will be expected to anticipate, document, and mitigate vulnerabilities not only in routine operations but also in emergency or shortage-prone contexts. This aligns the PQS with broader public health objectives, situating quality management as a bulwark against systemic healthcare risk.
International Harmonization and the ICH Q9(R1) Impact
Most significantly, the update explicitly references alignment with ICH Q9(R1) on Quality Risk Management, making harmonization with international best practice an explicit goal. This pushes organizations toward the global baseline for science- and risk-driven GMP.
The effect will be increased regulatory predictability for multinational manufacturers and heightened expectations for knowledge-handling and continuous improvement.
Summary Table: Draft vs. Current Chapter 1
Feature
Current Chapter 1 (2013)
Draft Chapter 1 (2025)
PQS Philosophy
Compliance/document control
Knowledge management & risk management
Risk Management
Implied, periodic
Embedded, real-time, evidence-based
ICH Q9 Alignment
Partial
Explicit, full alignment to Q9(R1)
Product Quality Review (PQR)
General guidance
Detailed, incl. grouped/low-batch
Supply Chain & Shortages
Minimal focus
Proactive risk, shortage prevention
Corrective/Preventive Action (CAPA)
System-oriented
Rooted in risk, causal prioritization
Lifecycle Integration
Weak
Strong, with embedded feedback
Operational Implications for Quality Leaders
The new Chapter 1 will demand a more dynamic, evidence-driven PQS, with robust mechanisms for knowledge transfer, risk-based priority setting, and system learning cycles. Technical writing, investigation reports, and CAPA logic will need to reference causal mechanisms and risk rationale explicitly—a marked shift from checklists to analytical narratives, aligning with the take-the-best causal reasoning discussed in your recent writings.
To prepare, organizations should:
Review and strengthen knowledge management assets
Embed risk assessment into the daily decision matrix—not just annual reviews
Foster investigative cultures that value causal specificity over exhaustive documentation
Reframe supply chain oversight as a continuous risk monitoring exercise
This systemic move, when enacted, will shift GMP thinking from historical compliance to forward-looking, adaptive quality management—an ambitious but necessary corrective for the challenges facing pharmaceutical manufacturing in 2025 and beyond.
The pharmaceutical industry has long operated under a fundamental epistemological fallacy that undermines our ability to truly understand the effectiveness of our quality systems. We celebrate zero deviations, zero recalls, zero adverse events, and zero regulatory observations as evidence that our systems are working. But a fundamental fact we tend to ignore is that we are confusing the absence of evidence with evidence of absence—a logical error that not only fails to prove effectiveness but actively impedes our ability to build more robust, science-based quality systems.
This challenge strikes at the heart of how we approach quality risk management. When our primary evidence of “success” is that nothing bad happened, we create unfalsifiable systems that can never truly be proven wrong.
The Philosophical Foundation: Falsifiability in Quality Risk Management
Karl Popper’s theory of falsification fundamentally challenges how we think about scientific validity. For Popper, the distinguishing characteristic of genuine scientific theories is not that they can be proven true, but that they can be proven false. A theory that cannot conceivably be refuted by any possible observation is not scientific—it’s metaphysical speculation.
Applied to quality risk management, this creates an uncomfortable truth: most of our current approaches to demonstrating system effectiveness are fundamentally unscientific. When we design quality systems around preventing negative outcomes and then use the absence of those outcomes as evidence of effectiveness, we create what Popper would call unfalsifiable propositions. No possible observation could ever prove our system ineffective as long as we frame effectiveness in terms of what didn’t happen.
Consider the typical pharmaceutical quality narrative: “Our manufacturing process is validated because we haven’t had any quality failures in twelve months.” This statement is unfalsifiable because it can always accommodate new information. If a failure occurs next month, we simply adjust our understanding of the system’s reliability without questioning the fundamental assumption that absence of failure equals validation. We might implement corrective actions, but we rarely question whether our original validation approach was capable of detecting the problems that eventually manifested.
Most of our current risk models are either highly predictive but untestable (making them useful for operational decisions but scientifically questionable) or neither predictive nor testable (making them primarily compliance exercises). The goal should be to move toward models are both scientifically rigorous and practically useful.
This philosophical foundation has practical implications for how we design and evaluate quality risk management systems. Instead of asking “How can we prevent bad things from happening?” we should be asking “How can we design systems that will fail in predictable ways when our underlying assumptions are wrong?” The first question leads to unfalsifiable defensive strategies; the second leads to falsifiable, scientifically valid approaches to quality assurance.
Why “Nothing Bad Happened” Isn’t Evidence of Effectiveness
The fundamental problem with using negative evidence to prove positive claims extends far beyond philosophical niceties, it creates systemic blindness that prevents us from understanding what actually drives quality outcomes. When we frame effectiveness in terms of absence, we lose the ability to distinguish between systems that work for the right reasons and systems that appear to work due to luck, external factors, or measurement limitations.
Scenario
Null Hypothesis
What Rejection Proves
What Non-Rejection Proves
Popperian Assessment
Traditional Efficacy Testing
No difference between treatment and control
Treatment is effective
Cannot prove effectiveness
Falsifiable and useful
Traditional Safety Testing
No increased risk
Treatment increases risk
Cannot prove safety
Unfalsifiable for safety
Absence of Events (Current)
No safety signal detected
Cannot prove anything
Cannot prove safety
Unfalsifiable
Non-inferiority Approach
Excess risk > acceptable margin
Treatment is acceptably safe
Cannot prove safety
Partially falsifiable
Falsification-Based Safety
Safety controls are inadequate
Current safety measures fail
Safety controls are adequate
Falsifiable and actionable
The table above demonstrates how traditional safety and effectiveness assessments fall into unfalsifiable categories. Traditional safety testing, for example, attempts to prove that something doesn’t increase risk, but this can never be definitively demonstrated—we can only fail to detect increased risk within the limitations of our study design. This creates a false confidence that may not be justified by the actual evidence.
The Sampling Illusion: When we observe zero deviations in a batch of 1000 units, we often conclude that our process is in control. But this conclusion conflates statistical power with actual system performance. With typical sampling strategies, we might have only 10% power to detect a 1% defect rate. The “zero observations” reflect our measurement limitations, not process capability.
The Survivorship Bias: Systems that appear effective may be surviving not because they’re well-designed, but because they haven’t yet encountered the conditions that would reveal their weaknesses. Our quality systems are often validated under ideal conditions and then extrapolated to real-world operations where different failure modes may dominate.
The Attribution Problem: When nothing bad happens, we attribute success to our quality systems without considering alternative explanations. Market forces, supplier improvements, regulatory changes, or simple random variation might be the actual drivers of observed outcomes.
Observable Outcome
Traditional Interpretation
Popperian Critique
What We Actually Know
Testable Alternative
Zero adverse events in 1000 patients
“The drug is safe”
Absence of evidence does not equal Evidence of absence
No events detected in this sample
Test limits of safety margin
Zero manufacturing deviations in 12 months
“The process is in control”
No failures observed does not equal a Failure-proof system
No deviations detected with current methods
Challenge process with stress conditions
Zero regulatory observations
“The system is compliant”
No findings does not equal No problems exist
No issues found during inspection
Audit against specific failure modes
Zero product recalls
“Quality is assured”
No recalls does not equal No quality issues
No quality failures reached market
Test recall procedures and detection
Zero patient complaints
“Customer satisfaction achieved”
No complaints does not equal No problems
No complaints received through channels
Actively solicit feedback mechanisms
This table illustrates how traditional interpretations of “positive” outcomes (nothing bad happened) fail to provide actionable knowledge. The Popperian critique reveals that these observations tell us far less than we typically assume, and the testable alternatives provide pathways toward more rigorous evaluation of system effectiveness.
The pharmaceutical industry’s reliance on these unfalsifiable approaches creates several downstream problems. First, it prevents genuine learning and improvement because we can’t distinguish effective interventions from ineffective ones. Second, it encourages defensive mindsets that prioritize risk avoidance over value creation. Third, it undermines our ability to make resource allocation decisions based on actual evidence of what works.
The Model Usefulness Problem: When Predictions Don’t Match Reality
George Box’s famous aphorism that “all models are wrong, but some are useful” provides a pragmatic framework for this challenge, but it doesn’t resolve the deeper question of how to determine when a model has crossed from “useful” to “misleading.” Popper’s falsifiability criterion offers one approach: useful models should make specific, testable predictions that could potentially be proven wrong by future observations.
The challenge in pharmaceutical quality management is that our models often serve multiple purposes that may be in tension with each other. Models used for regulatory submission need to demonstrate conservative estimates of risk to ensure patient safety. Models used for operational decision-making need to provide actionable insights for process optimization. Models used for resource allocation need to enable comparison of risks across different areas of the business.
When the same model serves all these purposes, it often fails to serve any of them well. Regulatory models become so conservative that they provide little guidance for actual operations. Operational models become so complex that they’re difficult to validate or falsify. Resource allocation models become so simplified that they obscure important differences in risk characteristics.
The solution isn’t to abandon modeling, but to be more explicit about the purpose each model serves and the criteria by which its usefulness should be judged. For regulatory purposes, conservative models that err on the side of safety may be appropriate even if they systematically overestimate risks. For operational decision-making, models should be judged primarily on their ability to correctly rank-order interventions by their impact on relevant outcomes. For scientific understanding, models should be designed to make falsifiable predictions that can be tested through controlled experiments or systematic observation.
Consider the example of cleaning validation, where we use models to predict the probability of cross-contamination between manufacturing campaigns. Traditional approaches focus on demonstrating that residual contamination levels are below acceptance criteria—essentially proving a negative. But this approach tells us nothing about the relative importance of different cleaning parameters, the margin of safety in our current procedures, or the conditions under which our cleaning might fail.
A more falsifiable approach would make specific predictions about how changes in cleaning parameters affect contamination levels. We might hypothesize that doubling the rinse time reduces contamination by 50%, or that certain product sequences create systematically higher contamination risks. These hypotheses can be tested and potentially falsified, providing genuine learning about the underlying system behavior.
From Defensive to Testable Risk Management
The evolution from defensive to testable risk management represents a fundamental shift in how we conceptualize quality systems. Traditional defensive approaches ask, “How can we prevent failures?” Testable approaches ask, “How can we design systems that fail predictably when our assumptions are wrong?” This shift moves us from unfalsifiable defensive strategies toward scientifically rigorous quality management.
This transition aligns with the broader evolution in risk thinking documented in ICH Q9(R1) and ISO 31000, which recognize risk as “the effect of uncertainty on objectives” where that effect can be positive, negative, or both. By expanding our definition of risk to include opportunities as well as threats, we create space for falsifiable hypotheses about system performance.
The integration of opportunity-based thinking with Popperian falsifiability creates powerful synergies. When we hypothesize that a particular quality intervention will not only reduce defects but also improve efficiency, we create multiple testable predictions. If the intervention reduces defects but doesn’t improve efficiency, we learn something important about the underlying system mechanics. If it improves efficiency but doesn’t reduce defects, we gain different insights. If it does neither, we discover that our fundamental understanding of the system may be flawed.
This approach requires a cultural shift from celebrating the absence of problems to celebrating the presence of learning. Organizations that embrace falsifiable quality management actively seek conditions that would reveal the limitations of their current systems. They design experiments to test the boundaries of their process capabilities. They view unexpected results not as failures to be explained away, but as opportunities to refine their understanding of system behavior.
The practical implementation of testable risk management involves several key elements:
Hypothesis-Driven Validation: Instead of demonstrating that processes meet specifications, validation activities should test specific hypotheses about process behavior. For example, rather than proving that a sterilization cycle achieves a 6-log reduction, we might test the hypothesis that cycle modifications affect sterility assurance in predictable ways. Instead of demonstrating that the CHO cell culture process consistently produces mAb drug substance meeting predetermined specifications, hypothesis-driven validation would test the specific prediction that maintaining pH at 7.0 ± 0.05 during the production phase will result in final titers that are 15% ± 5% higher than pH maintained at 6.9 ± 0.05, creating a falsifiable hypothesis that can be definitively proven wrong if the predicted titer improvement fails to materialize within the specified confidence intervals
Falsifiable Control Strategies: Control strategies should include specific predictions about how the system will behave under different conditions. These predictions should be testable and potentially falsifiable through routine monitoring or designed experiments.
Learning-Oriented Metrics: Key indicators should be designed to detect when our assumptions about system behavior are incorrect, not just when systems are performing within specification. Metrics that only measure compliance tell us nothing about the underlying system dynamics.
Proactive Stress Testing: Rather than waiting for problems to occur naturally, we should actively probe the boundaries of system performance through controlled stress conditions. This approach reveals failure modes before they impact patients while providing valuable data about system robustness.
Designing Falsifiable Quality Systems
The practical challenge of designing falsifiable quality systems requires a fundamental reconceptualization of how we approach quality assurance. Instead of building systems designed to prevent all possible failures, we need systems designed to fail in instructive ways when our underlying assumptions are incorrect.
This approach starts with making our assumptions explicit and testable. Traditional quality systems often embed numerous unstated assumptions about process behavior, material characteristics, environmental conditions, and human performance. These assumptions are rarely articulated clearly enough to be tested, making the systems inherently unfalsifiable. A falsifiable quality system makes these assumptions explicit and designs tests to evaluate their validity.
Consider the design of a typical pharmaceutical manufacturing process. Traditional approaches focus on demonstrating that the process consistently produces product meeting specifications under defined conditions. This demonstration typically involves process validation studies that show the process works under idealized conditions, followed by ongoing monitoring to detect deviations from expected performance.
A falsifiable approach would start by articulating specific hypotheses about what drives process performance. We might hypothesize that product quality is primarily determined by three critical process parameters, that these parameters interact in predictable ways, and that environmental variations within specified ranges don’t significantly impact these relationships. Each of these hypotheses can be tested and potentially falsified through designed experiments or systematic observation of process performance.
The key insight is that falsifiable quality systems are designed around testable theories of what makes quality systems effective, rather than around defensive strategies for preventing all possible problems. This shift enables genuine learning and continuous improvement because we can distinguish between interventions that work for the right reasons and those that appear to work for unknown or incorrect reasons.
Structured Hypothesis Formation: Quality requirements should be built around explicit hypotheses about cause-and-effect relationships in critical processes. These hypotheses should be specific enough to be tested and potentially falsified through systematic observation or experimentation.
Predictive Monitoring: Instead of monitoring for compliance with specifications, systems should monitor for deviations from predicted behavior. When predictions prove incorrect, this provides valuable information about the accuracy of our underlying process understanding.
Experimental Integration: Routine operations should be designed to provide ongoing tests of system hypotheses. Process changes, material variations, and environmental fluctuations should be treated as natural experiments that provide data about system behavior rather than disturbances to be minimized.
Failure Mode Anticipation: Quality systems should explicitly anticipate the ways failures might happen and design detection mechanisms for these failure modes. This proactive approach contrasts with reactive systems that only detect problems after they occur.
The Evolution of Risk Assessment: From Compliance to Science
The evolution of pharmaceutical risk assessment from compliance-focused activities to genuine scientific inquiry represents one of the most significant opportunities for improving quality outcomes. Traditional risk assessments often function primarily as documentation exercises designed to satisfy regulatory requirements rather than tools for genuine learning and improvement.
ICH Q9(R1) recognizes this limitation and calls for more scientifically rigorous approaches to quality risk management. The updated guidance emphasizes the need for risk assessments to be based on scientific knowledge and to provide actionable insights for quality improvement. This represents a shift away from checklist-based compliance activities toward hypothesis-driven scientific inquiry.
The integration of falsifiability principles with ICH Q9(R1) requirements creates opportunities for more rigorous and useful risk assessments. Instead of asking generic questions about what could go wrong, falsifiable risk assessments develop specific hypotheses about failure modes and design tests to evaluate these hypotheses. This approach provides more actionable insights while meeting regulatory expectations for systematic risk evaluation.
Consider the evolution of Failure Mode and Effects Analysis (FMEA) from a traditional compliance tool to a falsifiable risk assessment method. Traditional FMEA often devolves into generic lists of potential failures with subjective probability and impact assessments. The results provide limited insight because the assessments can’t be systematically tested or validated.
A falsifiable FMEA would start with specific hypotheses about failure mechanisms and their relationships to process parameters, material characteristics, or operational conditions. These hypotheses would be tested through historical data analysis, designed experiments, or systematic monitoring programs. The results would provide genuine insights into system behavior while creating a foundation for continuous improvement.
This evolution requires changes in how we approach several key risk assessment activities:
Hazard Identification: Instead of brainstorming all possible things that could go wrong, risk identification should focus on developing testable hypotheses about specific failure mechanisms and their triggers.
Risk Analysis: Probability and impact assessments should be based on testable models of system behavior rather than subjective expert judgment. When models prove inaccurate, this provides valuable information about the need to revise our understanding of system dynamics.
Risk Control: Control measures should be designed around testable theories of how interventions affect system behavior. The effectiveness of controls should be evaluated through systematic monitoring and periodic testing rather than assumed based on their implementation.
Risk Review: Risk review activities should focus on testing the accuracy of previous risk predictions and updating risk models based on new evidence. This creates a learning loop that continuously improves the quality of risk assessments over time.
Practical Framework for Falsifiable Quality Risk Management
The implementation of falsifiable quality risk management requires a systematic framework that integrates Popperian principles with practical pharmaceutical quality requirements. This framework must be sophisticated enough to generate genuine scientific insights while remaining practical for routine quality management activities.
The foundation of this framework rests on the principle that effective quality systems are built around testable theories of what drives quality outcomes. These theories should make specific predictions that can be evaluated through systematic observation, controlled experimentation, or historical data analysis. When predictions prove incorrect, this provides valuable information about the need to revise our understanding of system behavior.
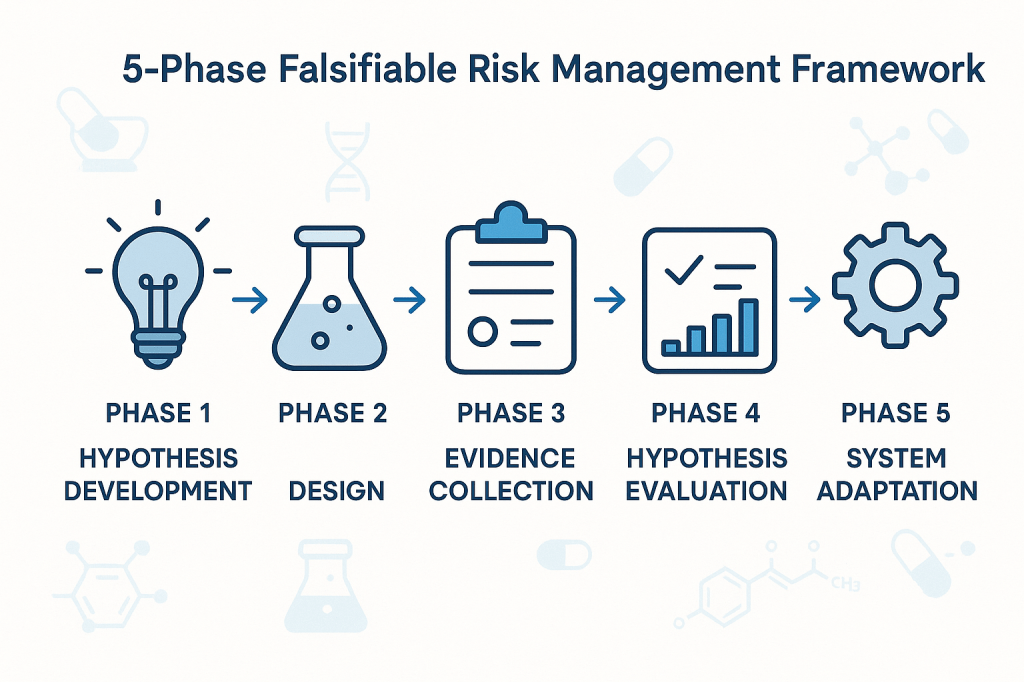
Phase 1: Hypothesis Development
The first phase involves developing specific, testable hypotheses about system behavior. These hypotheses should address fundamental questions about what drives quality outcomes in specific operational contexts. Rather than generic statements about quality risks, hypotheses should make specific predictions about relationships between process parameters, material characteristics, environmental conditions, and quality outcomes.
For example, instead of the generic hypothesis that “temperature variations affect product quality,” a falsifiable hypothesis might state that “temperature excursions above 25°C for more than 30 minutes during the mixing phase increase the probability of out-of-specification results by at least 20%.” This hypothesis is specific enough to be tested and potentially falsified through systematic data collection and analysis.
Phase 2: Experimental Design
The second phase involves designing systematic approaches to test the hypotheses developed in Phase 1. This might involve controlled experiments, systematic analysis of historical data, or structured monitoring programs designed to capture relevant data about hypothesis validity.
The key principle is that testing approaches should be capable of falsifying the hypotheses if they are incorrect. This requires careful attention to statistical power, measurement systems, and potential confounding factors that might obscure true relationships between variables.
Phase 3: Evidence Collection
The third phase focuses on systematic collection of evidence relevant to hypothesis testing. This evidence might come from designed experiments, routine monitoring data, or systematic analysis of historical performance. The critical requirement is that evidence collection should be structured around hypothesis testing rather than generic performance monitoring.
Evidence collection systems should be designed to detect when hypotheses are incorrect, not just when systems are performing within specifications. This requires more sophisticated approaches to data analysis and interpretation than traditional compliance-focused monitoring.
Phase 4: Hypothesis Evaluation
The fourth phase involves systematic evaluation of evidence against the hypotheses developed in Phase 1. This evaluation should follow rigorous statistical methods and should be designed to reach definitive conclusions about hypothesis validity whenever possible.
When hypotheses are falsified, this provides valuable information about the need to revise our understanding of system behavior. When hypotheses are supported by evidence, this provides confidence in our current understanding while suggesting areas for further testing and refinement.
Phase 5: System Adaptation
The final phase involves adapting quality systems based on the insights gained through hypothesis testing. This might involve modifying control strategies, updating risk assessments, or redesigning monitoring programs based on improved understanding of system behavior.
The critical principle is that system adaptations should be based on genuine learning about system behavior rather than reactive responses to compliance issues or external pressures. This creates a foundation for continuous improvement that builds cumulative knowledge about what drives quality outcomes.
Implementation Challenges
The transition to falsifiable quality risk management faces several practical challenges that must be addressed for successful implementation. These challenges range from technical issues related to experimental design and statistical analysis to cultural and organizational barriers that may resist more scientifically rigorous approaches to quality management.
Technical Challenges
The most immediate technical challenge involves designing falsifiable hypotheses that are relevant to pharmaceutical quality management. Many quality professionals have extensive experience with compliance-focused activities but limited experience with experimental design and hypothesis testing. This skills gap must be addressed through targeted training and development programs.
Statistical power represents another significant technical challenge. Many quality systems operate with very low baseline failure rates, making it difficult to design experiments with adequate power to detect meaningful differences in system performance. This requires sophisticated approaches to experimental design and may necessitate longer observation periods or larger sample sizes than traditionally used in quality management.
Measurement systems present additional challenges. Many pharmaceutical quality attributes are difficult to measure precisely, introducing uncertainty that can obscure true relationships between process parameters and quality outcomes. This requires careful attention to measurement system validation and uncertainty quantification.
Cultural and Organizational Challenges
Perhaps more challenging than technical issues are the cultural and organizational barriers to implementing more scientifically rigorous quality management approaches. Many pharmaceutical organizations have deeply embedded cultures that prioritize risk avoidance and compliance over learning and improvement.
The shift to falsifiable quality management requires cultural change that embraces controlled failure as a learning opportunity rather than something to be avoided at all costs. This represents a fundamental change in how many organizations think about quality management and may encounter significant resistance.
Regulatory relationships present additional organizational challenges. Many quality professionals worry that more rigorous scientific approaches to quality management might raise regulatory concerns or create compliance burdens. This requires careful communication with regulatory agencies to demonstrate that falsifiable approaches enhance rather than compromise patient safety.
Strategic Solutions
Successfully implementing falsifiable quality risk management requires strategic approaches that address both technical and cultural challenges. These solutions must be tailored to specific organizational contexts while maintaining scientific rigor and regulatory compliance.
Pilot Programs: Implementation should begin with carefully selected pilot programs in areas where falsifiable approaches can demonstrate clear value. These pilots should be designed to generate success stories that support broader organizational adoption while building internal capability and confidence.
Training and Development: Comprehensive training programs should be developed to build organizational capability in experimental design, statistical analysis, and hypothesis testing. These programs should be tailored to pharmaceutical quality contexts and should emphasize practical applications rather than theoretical concepts.
Regulatory Engagement: Proactive engagement with regulatory agencies should emphasize how falsifiable approaches enhance patient safety through improved understanding of system behavior. This communication should focus on the scientific rigor of the approach rather than on business benefits that might appear secondary to regulatory objectives.
Cultural Change Management: Systematic change management programs should address cultural barriers to embracing controlled failure as a learning opportunity. These programs should emphasize how falsifiable approaches support regulatory compliance and patient safety rather than replacing these priorities with business objectives.
Case Studies: Falsifiability in Practice
The practical application of falsifiable quality risk management can be illustrated through several case studies that demonstrate how Popperian principles can be integrated with routine pharmaceutical quality activities. These examples show how hypotheses can be developed, tested, and used to improve quality outcomes while maintaining regulatory compliance.
Case Study 1: Cleaning Validation Optimization
A biologics manufacturer was experiencing occasional cross-contamination events despite having validated cleaning procedures that consistently met acceptance criteria. Traditional approaches focused on demonstrating that cleaning procedures reduced contamination below specified limits, but provided no insight into the factors that occasionally caused this system to fail.
The falsifiable approach began with developing specific hypotheses about cleaning effectiveness. The team hypothesized that cleaning effectiveness was primarily determined by three factors: contact time with cleaning solution, mechanical action intensity, and rinse water temperature. They further hypothesized that these factors interacted in predictable ways and that current procedures provided a specific margin of safety above minimum requirements.
These hypotheses were tested through a designed experiment that systematically varied each cleaning parameter while measuring residual contamination levels. The results revealed that current procedures were adequate under ideal conditions but provided minimal margin of safety when multiple factors were simultaneously at their worst-case levels within specified ranges.
Based on these findings, the cleaning procedure was modified to provide greater margin of safety during worst-case conditions. More importantly, ongoing monitoring was redesigned to test the continued validity of the hypotheses about cleaning effectiveness rather than simply verifying compliance with acceptance criteria.
Case Study 2: Process Control Strategy Development
A pharmaceutical manufacturer was developing a control strategy for a new manufacturing process. Traditional approaches would have focused on identifying critical process parameters and establishing control limits based on process validation studies. Instead, the team used a falsifiable approach that started with explicit hypotheses about process behavior.
The team hypothesized that product quality was primarily controlled by the interaction between temperature and pH during the reaction phase, that these parameters had linear effects on product quality within the normal operating range, and that environmental factors had negligible impact on these relationships.
These hypotheses were tested through systematic experimentation during process development. The results confirmed the importance of the temperature-pH interaction but revealed nonlinear effects that weren’t captured in the original hypotheses. More importantly, environmental humidity was found to have significant effects on process behavior under certain conditions.
The control strategy was designed around the revised understanding of process behavior gained through hypothesis testing. Ongoing process monitoring was structured to continue testing key assumptions about process behavior rather than simply detecting deviations from target conditions.
Case Study 3: Supplier Quality Management
A biotechnology company was managing quality risks from a critical raw material supplier. Traditional approaches focused on incoming inspection and supplier auditing to verify compliance with specifications and quality system requirements. However, occasional quality issues suggested that these approaches weren’t capturing all relevant quality risks.
The falsifiable approach started with specific hypotheses about what drove supplier quality performance. The team hypothesized that supplier quality was primarily determined by their process control during critical manufacturing steps, that certain environmental conditions increased the probability of quality issues, and that supplier quality system maturity was predictive of long-term quality performance.
These hypotheses were tested through systematic analysis of supplier quality data, enhanced supplier auditing focused on specific process control elements, and structured data collection about environmental conditions during material manufacturing. The results revealed that traditional quality system assessments were poor predictors of actual quality performance, but that specific process control practices were strongly predictive of quality outcomes.
The supplier management program was redesigned around the insights gained through hypothesis testing. Instead of generic quality system requirements, the program focused on specific process control elements that were demonstrated to drive quality outcomes. Supplier performance monitoring was structured around testing continued validity of the relationships between process control and quality outcomes.
Measuring Success in Falsifiable Quality Systems
The evaluation of falsifiable quality systems requires fundamentally different approaches to performance measurement than traditional compliance-focused systems. Instead of measuring the absence of problems, we need to measure the presence of learning and the accuracy of our predictions about system behavior.
Traditional quality metrics focus on outcomes: defect rates, deviation frequencies, audit findings, and regulatory observations. While these metrics remain important for regulatory compliance and business performance, they provide limited insight into whether our quality systems are actually effective or merely lucky. Falsifiable quality systems require additional metrics that evaluate the scientific validity of our approach to quality management.
Predictive Accuracy Metrics
The most direct measure of a falsifiable quality system’s effectiveness is the accuracy of its predictions about system behavior. These metrics evaluate how well our hypotheses about quality system behavior match observed outcomes. High predictive accuracy suggests that we understand the underlying drivers of quality outcomes. Low predictive accuracy indicates that our understanding needs refinement.
Predictive accuracy metrics might include the percentage of process control predictions that prove correct, the accuracy of risk assessments in predicting actual quality issues, or the correlation between predicted and observed responses to process changes. These metrics provide direct feedback about the validity of our theoretical understanding of quality systems.
Learning Rate Metrics
Another important category of metrics evaluates how quickly our understanding of quality systems improves over time. These metrics measure the rate at which falsified hypotheses lead to improved system performance or more accurate predictions. High learning rates indicate that the organization is effectively using falsifiable approaches to improve quality outcomes.
Learning rate metrics might include the time required to identify and correct false assumptions about system behavior, the frequency of successful process improvements based on hypothesis testing, or the rate of improvement in predictive accuracy over time. These metrics evaluate the dynamic effectiveness of falsifiable quality management approaches.
Hypothesis Quality Metrics
The quality of hypotheses generated by quality risk management processes represents another important performance dimension. High-quality hypotheses are specific, testable, and relevant to important quality outcomes. Poor-quality hypotheses are vague, untestable, or focused on trivial aspects of system performance.
Hypothesis quality can be evaluated through structured peer review processes, assessment of testability and specificity, and evaluation of relevance to critical quality attributes. Organizations with high-quality hypothesis generation processes are more likely to gain meaningful insights from their quality risk management activities.
System Robustness Metrics
Falsifiable quality systems should become more robust over time as learning accumulates and system understanding improves. Robustness can be measured through the system’s ability to maintain performance despite variations in operating conditions, changes in materials or equipment, or other sources of uncertainty.
Robustness metrics might include the stability of process performance across different operating conditions, the effectiveness of control strategies under stress conditions, or the system’s ability to detect and respond to emerging quality risks. These metrics evaluate whether falsifiable approaches actually lead to more reliable quality systems.
Regulatory Implications and Opportunities
The integration of falsifiable principles with pharmaceutical quality risk management creates both challenges and opportunities in regulatory relationships. While some regulatory agencies may initially view scientific approaches to quality management with skepticism, the ultimate result should be enhanced regulatory confidence in quality systems that can demonstrate genuine understanding of what drives quality outcomes.
The key to successful regulatory engagement lies in emphasizing how falsifiable approaches enhance patient safety rather than replacing regulatory compliance with business optimization. Regulatory agencies are primarily concerned with patient safety and product quality. Falsifiable quality systems support these objectives by providing more rigorous and reliable approaches to ensuring quality outcomes.
Enhanced Regulatory Submissions
Regulatory submissions based on falsifiable quality systems can provide more compelling evidence of system effectiveness than traditional compliance-focused approaches. Instead of demonstrating that systems meet minimum requirements, falsifiable approaches can show genuine understanding of what drives quality outcomes and how systems will behave under different conditions.
This enhanced evidence can support regulatory flexibility in areas such as process validation, change control, and ongoing monitoring requirements. Regulatory agencies may be willing to accept risk-based approaches to these activities when they’re supported by rigorous scientific evidence rather than generic compliance activities.
Proactive Risk Communication
Falsifiable quality systems enable more proactive and meaningful communication with regulatory agencies about quality risks and mitigation strategies. Instead of reactive communication about compliance issues, organizations can engage in scientific discussions about system behavior and improvement strategies.
This proactive communication can build regulatory confidence in organizational quality management capabilities while providing opportunities for regulatory agencies to provide input on scientific approaches to quality improvement. The result should be more collaborative regulatory relationships based on shared commitment to scientific rigor and patient safety.
Regulatory Science Advancement
The pharmaceutical industry’s adoption of more scientifically rigorous approaches to quality management can contribute to the advancement of regulatory science more broadly. Regulatory agencies benefit from industry innovations in risk assessment, process understanding, and quality assurance methods.
Organizations that successfully implement falsifiable quality risk management can serve as case studies for regulatory guidance development and can provide evidence for the effectiveness of science-based approaches to quality assurance. This contribution to regulatory science advancement creates value that extends beyond individual organizational benefits.
Toward a More Scientific Quality Culture
The long-term vision for falsifiable quality risk management extends beyond individual organizational implementations to encompass fundamental changes in how the pharmaceutical industry approaches quality assurance. This vision includes more rigorous scientific approaches to quality management, enhanced collaboration between industry and regulatory agencies, and continuous advancement in our understanding of what drives quality outcomes.
Industry-Wide Learning Networks
One promising direction involves the development of industry-wide learning networks that share insights from falsifiable quality management implementations. These networks facilitate collaborative hypothesis testing, shared learning from experimental results, and development of common methodologies for scientific approaches to quality assurance.
Such networks accelerate the advancement of quality science while maintaining appropriate competitive boundaries. Organizations should share methodological insights and general findings while protecting proprietary information about specific processes or products. The result would be faster advancement in quality management science that benefits the entire industry.
Advanced Analytics Integration
The integration of advanced analytics and machine learning techniques with falsifiable quality management approaches represents another promising direction. These technologies can enhance our ability to develop testable hypotheses, design efficient experiments, and analyze complex datasets to evaluate hypothesis validity.
Machine learning approaches are particularly valuable for identifying patterns in complex quality datasets that might not be apparent through traditional analysis methods. However, these approaches must be integrated with falsifiable frameworks to ensure that insights can be validated and that predictive models can be systematically tested and improved.
Regulatory Harmonization
The global harmonization of regulatory approaches to science-based quality management represents a significant opportunity for advancing patient safety and regulatory efficiency. As individual regulatory agencies gain experience with falsifiable quality management approaches, there are opportunities to develop harmonized guidance that supports consistent global implementation.
ICH Q9(r1) was a great step. I would love to see continued work in this area.
Embracing the Discomfort of Scientific Rigor
The transition from compliance-focused to scientifically rigorous quality risk management represents more than a methodological change—it requires fundamentally rethinking how we approach quality assurance in pharmaceutical manufacturing. By embracing Popper’s challenge that genuine scientific theories must be falsifiable, we move beyond the comfortable but ultimately unhelpful world of proving negatives toward the more demanding but ultimately more rewarding world of testing positive claims about system behavior.
The effectiveness paradox that motivates this discussion—the problem of determining what works when our primary evidence is that “nothing bad happened”—cannot be resolved through better compliance strategies or more sophisticated documentation. It requires genuine scientific inquiry into the mechanisms that drive quality outcomes. This inquiry must be built around testable hypotheses that can be proven wrong, not around defensive strategies that can always accommodate any possible outcome.
The practical implementation of falsifiable quality risk management is not without challenges. It requires new skills, different cultural approaches, and more sophisticated methodologies than traditional compliance-focused activities. However, the potential benefits—genuine learning about system behavior, more reliable quality outcomes, and enhanced regulatory confidence—justify the investment required for successful implementation.
Perhaps most importantly, the shift to falsifiable quality management moves us toward a more honest assessment of what we actually know about quality systems versus what we merely assume or hope to be true. This honesty is uncomfortable but essential for building quality systems that genuinely serve patient safety rather than organizational comfort.
The question is not whether pharmaceutical quality management will eventually embrace more scientific approaches—the pressures of regulatory evolution, competitive dynamics, and patient safety demands make this inevitable. The question is whether individual organizations will lead this transition or be forced to follow. Those that embrace the discomfort of scientific rigor now will be better positioned to thrive in a future where quality management is evaluated based on genuine effectiveness rather than compliance theater.
As we continue to navigate an increasingly complex regulatory and competitive environment, the organizations that master the art of turning uncertainty into testable knowledge will be best positioned to deliver consistent quality outcomes while maintaining the flexibility needed for innovation and continuous improvement. The integration of Popperian falsifiability with modern quality risk management provides a roadmap for achieving this mastery while maintaining the rigorous standards our industry demands.
The path forward requires courage to question our current assumptions, discipline to design rigorous tests of our theories, and wisdom to learn from both our successes and our failures. But for those willing to embrace these challenges, the reward is quality systems that are not only compliant but genuinely effective. Systems that we can defend not because they’ve never been proven wrong, but because they’ve been proven right through systematic, scientific inquiry.
If there is one section that serves as the philosophical and operational backbone for everything else in the new regulation, it’s Section 4: Risk Management. This section embodies current regulatory thinking on how risk management, in light of the recent ICH Q9 (R1) is the scientific methodology that transforms how we think about, design, validate, and operate s in GMP environments.
Section 4 represents the regulatory codification of what quality professionals have long advocated: that every decision about computerized systems, from initial selection through operational oversight to eventual decommissioning, must be grounded in rigorous, documented, and scientifically defensible risk assessment. But more than that, it establishes quality risk management as the living nervous system of digital compliance, continuously sensing, evaluating, and responding to threats and opportunities throughout the system lifecycle.
For organizations that have treated risk management as a checkbox exercise or a justification for doing less validation, Section 4 delivers a harsh wake-up call. The new requirements don’t just elevate risk management to regulatory mandate—they transform it into the primary lens through which all computerized system activities must be viewed, planned, executed, and continuously improved.
The Philosophical Revolution: From Optional Framework to Mandatory Foundation
The transformation between the current Annex 11’s brief mention of risk management and Section 4’s comprehensive requirements represents more than regulatory updating—it reflects a fundamental shift in how regulators view the relationship between risk assessment and system control. Where the 2011 version offered generic guidance about applying risk management “throughout the lifecycle,” Section 4 establishes specific, measurable, and auditable requirements that make risk management the definitive basis for all computerized system decisions.
Section 4.1 opens with an unambiguous statement that positions quality risk management as the foundation of system lifecycle management: “Quality Risk Management (QRM) should be applied throughout the lifecycle of a computerised system considering any possible impact on product quality, patient safety or data integrity.” This language moves beyond the permissive “should consider” of the old regulation to establish QRM as the mandatory framework through which all system activities must be filtered.
The explicit connection to ICH Q9(R1) in Section 4.2 represents a crucial evolution. By requiring that “risks associated with the use of computerised systems in GMP activities should be identified and analysed according to an established procedure” and specifically referencing “examples of risk management methods and tools can be found in ICH Q9 (R1),” the regulation transforms ICH Q9 from guidance into regulatory requirement. Organizations can no longer treat ICH Q9 principles as aspirational best practices—they become the enforceable standard for pharmaceutical risk management.
This integration creates powerful synergies between pharmaceutical quality system requirements and computerized system validation. Risk assessments conducted under Section 4 must align with broader ICH Q9 principles while addressing the specific challenges of digital systems, cloud services, and automated processes. The result is a comprehensive risk management framework that bridges traditional pharmaceutical operations with modern digital infrastructure.
The requirement in Section 4.3 that “validation strategy and effort should be determined based on the intended use of the system and potential risks to product quality, patient safety and data integrity” establishes risk assessment as the definitive driver of validation scope and approach. This eliminates the historical practice of using standardized validation templates regardless of system characteristics or applying uniform validation approaches across diverse system types.
Under Section 4, every validation decision—from the depth of testing required to the frequency of periodic reviews—must be traceable to specific risk assessments that consider the unique characteristics of each system and its role in GMP operations. This approach rewards organizations that invest in comprehensive risk assessment while penalizing those that rely on generic, one-size-fits-all validation approaches.
Risk-Based System Design: Architecture Driven by Assessment
Perhaps the most transformative aspect of Section 4 is found in Section 4.4, which requires that “risks associated with the use of computerised systems in GMP activities should be mitigated and brought down to an acceptable level, if possible, by modifying processes or system design.” This requirement positions risk assessment as a primary driver of system architecture rather than simply a validation planning tool.
The language “modifying processes or system design” establishes a hierarchy of risk control that prioritizes prevention over detection. Rather than accepting inherent system risks and compensating through enhanced testing or operational controls, Section 4 requires organizations to redesign systems and processes to eliminate or minimize risks at their source. This approach aligns with fundamental safety engineering principles while ensuring that risk mitigation is built into system architecture rather than layered on top.
The requirement that “the outcome of the risk management process should result in the choice of an appropriate computerised system architecture and functionality” makes risk assessment the primary criterion for system selection and configuration. Organizations can no longer choose systems based purely on cost, vendor relationships, or technical preferences—they must demonstrate that system architecture aligns with risk assessment outcomes and provides appropriate risk mitigation capabilities.
This approach particularly impacts cloud system implementations, SaaS platform selections, and integrated system architectures where risk assessment must consider not only individual system capabilities but also the risk implications of system interactions, data flows, and shared infrastructure. Organizations must demonstrate that their chosen architecture provides adequate risk control across the entire integrated environment.
The emphasis on system design modification as the preferred risk mitigation approach will drive significant changes in vendor selection criteria and system specification processes. Vendors that can demonstrate built-in risk controls and flexible architecture will gain competitive advantages over those that rely on customers to implement risk mitigation through operational procedures or additional validation activities.
Data Integrity Risk Assessment: Scientific Rigor Applied to Information Management
Section 4.5 introduces one of the most sophisticated requirements in the entire draft regulation: “Quality risk management principles should be used to assess the criticality of data to product quality, patient safety and data integrity, the vulnerability of data to deliberate or indeliberate alteration, deletion or loss, and the likelihood of detection of such actions.”
This requirement transforms data integrity from a compliance concept into a systematic risk management discipline. Organizations must assess not only what data is critical but also how vulnerable that data is to compromise and how likely they are to detect integrity failures. This three-dimensional risk assessment approach—criticality, vulnerability, and detectability—provides a scientific framework for prioritizing data protection efforts and designing appropriate controls.
The distinction between “deliberate or indeliberate” data compromise acknowledges that modern data integrity threats encompass both malicious attacks and innocent errors. Risk assessments must consider both categories and design controls that address the full spectrum of potential data integrity failures. This approach requires organizations to move beyond traditional access control and audit trail requirements to consider the full range of technical, procedural, and human factors that could compromise data integrity.
The requirement to assess “likelihood of detection” introduces a crucial element often missing from traditional data integrity approaches. Organizations must evaluate not only how to prevent data integrity failures but also how quickly and reliably they can detect failures that occur despite preventive controls. This assessment drives requirements for monitoring systems, audit trail analysis capabilities, and incident detection procedures that can identify data integrity compromises before they impact product quality or patient safety.
This risk-based approach to data integrity creates direct connections between Section 4 and other draft Annex 11 requirements, particularly Section 10 (Handling of Data), Section 11 (Identity and Access Management), and Section 12 (Audit Trails). Risk assessments conducted under Section 4 drive the specific requirements for data input verification, access controls, and audit trail monitoring implemented through other sections.
Lifecycle Risk Management: Dynamic Assessment in Digital Environments
The lifecycle approach required by Section 4 acknowledges that computerized systems exist in dynamic environments where risks evolve continuously due to technology changes, process modifications, security threats, and operational experience. Unlike traditional validation approaches that treat risk assessment as a one-time activity during system implementation, Section 4 requires ongoing risk evaluation and response throughout the system lifecycle.
This dynamic approach particularly impacts cloud-based systems and SaaS platforms where underlying infrastructure, security controls, and functional capabilities change regularly without direct customer involvement. Organizations must establish procedures for evaluating the risk implications of vendor-initiated changes and updating their risk assessments and control strategies accordingly.
The lifecycle risk management approach also requires integration with change control processes, periodic review activities, and incident management procedures. Every significant system change must trigger risk reassessment to ensure that new risks are identified and appropriate controls are implemented. This creates a feedback loop where operational experience informs risk assessment updates, which in turn drive control system improvements and validation strategy modifications.
Organizations implementing Section 4 requirements must develop capabilities for continuous risk monitoring that can detect emerging threats, changing system characteristics, and evolving operational patterns that might impact risk assessments. This requires investment in risk management tools, monitoring systems, and analytical capabilities that extend beyond traditional validation and quality assurance functions.
Integration with Modern Risk Management Methodologies
The explicit reference to ICH Q9(R1) in Section 4.2 creates direct alignment between computerized system risk management and the broader pharmaceutical quality risk management framework. This integration ensures that computerized system risk assessments contribute to overall product and process risk understanding while benefiting from the sophisticated risk management methodologies developed for pharmaceutical operations.
ICH Q9(R1)’s emphasis on managing and minimizing subjectivity in risk assessment becomes particularly important for computerized system applications where technical complexity can obscure risk evaluation. Organizations must implement risk assessment procedures that rely on objective data, established methodologies, and cross-functional expertise rather than individual opinions or vendor assertions.
The ICH Q9(R1) toolkit—including Failure Mode and Effects Analysis (FMEA), Hazard Analysis and Critical Control Points (HACCP), and Fault Tree Analysis (FTA)—provides proven methodologies for systematic risk identification and assessment that can be applied to computerized system environments. Section 4’s reference to these tools establishes them as acceptable approaches for meeting regulatory requirements while providing flexibility for organizations to choose methodologies appropriate to their specific circumstances.
The integration with ICH Q9(R1) also emphasizes the importance of risk communication throughout the organization and with external stakeholders including suppliers, regulators, and business partners. Risk assessment results must be communicated effectively to drive appropriate decision-making at all organizational levels and ensure that risk mitigation strategies are understood and implemented consistently.
Operational Implementation: Transforming Risk Assessment from Theory to Practice
Implementing Section 4 requirements effectively requires organizations to develop sophisticated risk management capabilities that extend far beyond traditional validation and quality assurance functions. The requirement for “established procedures” means that risk assessment cannot be ad hoc or inconsistent—organizations must develop repeatable, documented methodologies that produce reliable and auditable results.
The procedures must address risk identification methods that can systematically evaluate the full range of potential threats to computerized systems including technical failures, security breaches, data integrity compromises, supplier issues, and operational errors. Risk identification must consider both current system states and future scenarios including planned changes, emerging threats, and evolving operational requirements.
Risk analysis procedures must provide quantitative or semi-quantitative methods for evaluating risk likelihood and impact across the three critical dimensions specified in Section 4.1: product quality, patient safety, and data integrity. This analysis must consider the interconnected nature of modern computerized systems where risks in one system or component can cascade through integrated environments to impact multiple processes and outcomes.
Risk evaluation procedures must establish criteria for determining acceptable risk levels and identifying risks that require mitigation. These criteria must align with organizational risk tolerance, regulatory expectations, and business objectives while providing clear guidance for risk-based decision making throughout the system lifecycle.
Risk mitigation procedures must prioritize design and process modifications over operational controls while ensuring that all risk mitigation strategies are evaluated for effectiveness and maintained throughout the system lifecycle. Organizations must develop capabilities for implementing system architecture changes, process redesign, and operational control enhancements based on risk assessment outcomes.
Technology and Tool Requirements for Effective Risk Management
Section 4’s emphasis on systematic, documented, and traceable risk management creates significant requirements for technology tools and platforms that can support sophisticated risk assessment and management processes. Organizations must invest in risk management systems that can capture, analyze, and track risks throughout complex system lifecycles while maintaining traceability to validation activities, change control processes, and operational decisions.
Risk assessment tools must support the multi-dimensional analysis required by Section 4, including product quality impacts, patient safety implications, and data integrity vulnerabilities. These tools must accommodate the dynamic nature of computerized system environments where risks evolve continuously due to technology changes, process modifications, and operational experience.
Integration with existing quality management systems, validation platforms, and operational monitoring tools becomes essential for maintaining consistency between risk assessments and other quality activities. Organizations must ensure that risk assessment results drive validation planning, change control decisions, and operational monitoring strategies while receiving feedback from these activities to update and improve risk assessments.
Documentation and traceability requirements create needs for sophisticated document management and workflow systems that can maintain relationships between risk assessments, system specifications, validation protocols, and operational procedures. Organizations must demonstrate clear traceability from risk identification through mitigation implementation and effectiveness verification.
Regulatory Expectations and Inspection Implications
Section 4’s comprehensive risk management requirements fundamentally change regulatory inspection dynamics by establishing risk assessment as the foundation for evaluating all computerized system compliance activities. Inspectors will expect to see documented, systematic, and scientifically defensible risk assessments that drive all system-related decisions from initial selection through ongoing operation.
The integration with ICH Q9(R1) provides inspectors with established criteria for evaluating risk management effectiveness including assessment methodology adequacy, stakeholder involvement appropriateness, and decision-making transparency. Organizations must demonstrate that their risk management processes meet ICH Q9(R1) standards while addressing the specific challenges of computerized system environments.
Risk-based validation approaches will receive increased scrutiny as inspectors evaluate whether validation scope and depth align appropriately with documented risk assessments. Organizations that cannot demonstrate clear traceability between risk assessments and validation activities will face significant compliance challenges regardless of validation execution quality.
The emphasis on system design and process modification as preferred risk mitigation strategies means that inspectors will evaluate whether organizations have adequately considered architectural and procedural alternatives to operational controls. Simply implementing extensive operational procedures to manage inherent system risks may no longer be considered adequate risk mitigation.
Ongoing risk management throughout the system lifecycle will become a key inspection focus as regulators evaluate whether organizations maintain current risk assessments and adjust control strategies based on operational experience, technology changes, and emerging threats. Static risk assessments that remain unchanged throughout system operation will be viewed as inadequate regardless of initial quality.
Strategic Implications for Pharmaceutical Operations
Section 4’s requirements represent a strategic inflection point for pharmaceutical organizations as they transition from compliance-driven computerized system approaches to risk-based digital strategies. Organizations that excel at implementing Section 4 requirements will gain competitive advantages through more effective system selection, optimized validation strategies, and superior operational risk management.
The emphasis on risk-driven system architecture creates opportunities for organizations to differentiate themselves through superior system design and integration strategies. Organizations that can demonstrate sophisticated risk assessment capabilities and implement appropriate system architectures will achieve better operational outcomes while reducing compliance costs and regulatory risks.
Risk-based validation approaches enabled by Section 4 provide opportunities for more efficient resource allocation and faster system implementation timelines. Organizations that invest in comprehensive risk assessment capabilities can focus validation efforts on areas of highest risk while reducing unnecessary validation activities for lower-risk system components and functions.
The integration with ICH Q9(R1) creates opportunities for pharmaceutical organizations to leverage their existing quality risk management capabilities for computerized system applications while enhancing overall organizational risk management maturity. Organizations can achieve synergies between product quality risk management and system risk management that improve both operational effectiveness and regulatory compliance.
Future Evolution and Continuous Improvement
Section 4’s lifecycle approach to risk management positions organizations for continuous improvement in risk assessment and mitigation capabilities as they gain operational experience and encounter new challenges. The requirement for ongoing risk evaluation creates feedback loops that enable organizations to refine their risk management approaches based on real-world performance and emerging best practices.
The dynamic nature of computerized system environments means that risk management capabilities must evolve continuously to address new technologies, changing threats, and evolving operational requirements. Organizations that establish robust risk management foundations under Section 4 will be better positioned to adapt to future regulatory changes and technology developments.
The integration with broader pharmaceutical quality systems creates opportunities for organizations to develop comprehensive risk management capabilities that span traditional manufacturing operations and modern digital infrastructure. This integration enables more sophisticated risk assessment and mitigation strategies that consider the full range of factors affecting product quality, patient safety, and data integrity.
Organizations that embrace Section 4’s requirements as strategic capabilities rather than compliance obligations will build sustainable competitive advantages through superior risk management that enables more effective system selection, optimized operational strategies, and enhanced regulatory relationships.
The Foundation for Digital Transformation
Section 4 ultimately serves as the scientific foundation for pharmaceutical digital transformation by providing the risk management framework necessary to evaluate, implement, and operate sophisticated computerized systems with appropriate confidence and control. The requirement for systematic, documented, and traceable risk assessment provides the methodology necessary to navigate the complex risk landscapes of modern pharmaceutical operations.
The emphasis on risk-driven system design creates the foundation for implementing advanced technologies including artificial intelligence, machine learning, and automated process control with appropriate risk understanding and mitigation. Organizations that master Section 4’s requirements will be positioned to leverage these technologies effectively while maintaining regulatory compliance and operational control.
The lifecycle approach to risk management provides the framework necessary to manage the continuous evolution of computerized systems in dynamic business and regulatory environments. Organizations that implement Section 4 requirements effectively will build the capabilities necessary to adapt continuously to changing circumstances while maintaining consistent risk management standards.
Section 4 represents more than regulatory compliance—it establishes the scientific methodology that enables pharmaceutical organizations to harness the full potential of digital technologies while maintaining the rigorous risk management standards essential for protecting product quality, patient safety, and data integrity. Organizations that embrace this transformation will lead the industry’s evolution toward more sophisticated, efficient, and effective pharmaceutical operations.
Requirement Area
Draft Annex 11 Section 4 (2025)
Current Annex 11 (2011)
ICH Q9(R1) 2023
Implementation Impact
Lifecycle Application
QRM applied throughout entire lifecycle considering product quality, patient safety, data integrity
Risk management throughout lifecycle considering patient safety, data integrity, product quality
Quality risk management throughout product lifecycle
Requires continuous risk assessment processes rather than one-time validation activities
Risk Assessment Focus
Risks identified and analyzed per established procedure with ICH Q9(R1) methods
Risk assessment should consider patient safety, data integrity, product quality
Systematic risk identification, analysis, and evaluation
Mandates systematic procedures using proven methodologies rather than ad hoc approaches
Validation Strategy
Validation strategy and effort determined based on intended use and potential risks
Validation extent based on justified and documented risk assessment
Risk-based approach to validation and control strategies
Links validation scope directly to risk assessment outcomes, potentially reducing or increasing validation burden
Risk Mitigation
Risks mitigated to acceptable level through process/system design modifications
Risk mitigation not explicitly detailed
Risk control through reduction and acceptance strategies
Prioritizes system design changes over operational controls, potentially requiring architecture modifications
Data Integrity Risk
QRM principles assess data criticality, vulnerability, detection likelihood
Data integrity risk mentioned but not detailed
Data integrity risks as part of overall quality risk assessment
Requires sophisticated three-dimensional risk assessment for all data management activities
Documentation Requirements
Documented risk assessments required for all computerized systems
Risk assessment should be justified and documented
Documented, transparent, and reproducible risk management processes
Elevates documentation standards and requires traceability throughout system lifecycle
Integration with QRM
Fully integrated with ICH Q9(R1) quality risk management principles
General risk management principles
Core principle of pharmaceutical quality system
Creates mandatory alignment between system and product risk management activities
Ongoing Risk Review
Risk review required for changes and incidents throughout lifecycle
Risk review not explicitly required
Regular risk review based on new knowledge and experience
Establishes continuous risk monitoring as operational requirement rather than periodic activity
The environment for commissioning, qualification, and validation (CQV) professionals remains defined by persistent challenges. Rapid technological advancements—most notably in artificial intelligence, machine learning, and automation—are constantly reshaping the expectations for validation. Compliance requirements are in frequent flux as agencies modernize guidance, while the complexity of novel biologics and therapies demands ever-higher standards of sterility, traceability, and process control. The shift towards digital systems has introduced significant hurdles in data management and integration, often stretching already limited resources. At the same time, organizations are expected to fully embrace risk-based, science-first approaches, which require new methodologies and skills. Finally, true validation now hinges on effective collaboration and knowledge-sharing among increasingly cross-functional and global teams.
Overlaying these challenges, three major regulatory paradigm shifts are transforming the expectations around risk management, contamination control, and data integrity. Data integrity in particular has become an international touchpoint. Since the landmark PIC/S guidance in 2021 and matching World Health Organization updates, agencies have made it clear that trustworthy, accurate, and defendable data—whether paper-based or digital—are the foundation of regulatory confidence. Comprehensive data governance, end-to-end traceability, and robust documentation are now all non-negotiable.
Contamination control is experiencing its own revolution. The August 2023 overhaul of EU GMP Annex 1 set a new benchmark for sterile manufacturing. The core concept, the Contamination Control Strategy (CCS), formalizes expectations: every manufacturer must systematically identify, map, and control contamination risks across the entire product lifecycle. From supply chain vigilance to environmental monitoring, regulators are pushing for a proactive, science-driven, and holistic approach, far beyond previous practices that too often relied on reactive measures. We this reflected in recent USP drafts as well.
Quality risk management (QRM) also has a new regulatory backbone. The ICH Q9(R1) revision, finalized in 2023, addresses long-standing shortcomings—particularly subjectivity and lack of consistency—in how risks are identified and managed. The European Medicines Agency’s ongoing revision of EudraLex Chapter 1, now aiming for finalization in 2026, will further require organizations to embed preventative, science-based risk management within globalized and complex supply chain operations. Modern products and supply webs simply cannot be managed with last-generation compliance thinking.
The EU Digital Modernization: Chapter 4, Annex 11, and Annex 22
With the rapid digitalization of pharma, the European Union has embarked on an ambitious modernization of its GMP framework. At the heart of these changes are the upcoming revisions to Chapter 4 (Documentation), Annex 11 (Computerised Systems), and the anticipated implementation of Annex 22 (Artificial Intelligence).
Chapter 4—Documentation is being thoroughly updated in parallel with Annex 11. The current chapter, which governs all aspects of documentation in GMP environments, was last revised in 2011. Its modernization is a direct response to the prevalence of digital tools—electronic records, digital signatures, and interconnected documentation systems. The revised Chapter 4 is expected to provide much clearer requirements for the management, review, retention, and security of both paper and electronic records, ensuring that information flows align seamlessly with the increasingly digital processes described in Annex 11. Together, these updates will enable companies to phase out paper where possible, provided electronic systems are validated, auditable, and secure.
Annex 11—Computerised Systems will see its most significant overhaul since the dawn of digital pharma. The new guidance, scheduled for publication and adoption in 2026, directly addresses areas that the previous version left insufficiently covered. The scope now embraces the tectonic shift toward AI, machine learning, cloud-based services, agile project management, and advanced digital workflows. For instance, close attention is being paid to the robustness of electronic signatures, demanding multi-factor authentication, time-zoned audit trails, and explicit provisions for non-repudiation. Hybrid (wet-ink/digital) records will only be acceptable if they can demonstrate tamper-evidence via hashes or equivalent mechanisms. Especially significant is the regulation of “open systems” such as SaaS and cloud platforms. Here, organizations can no longer rely on traditional username/password models; instead, compliance with standards like eIDAS for trusted digital providers is expected, with more of the technical compliance burden shifting onto certified digital partners.
The new Annex 11 also calls for enhanced technical controls throughout computerized systems, proportional risk management protocols for new technologies, and a far greater emphasis on continuous supplier oversight and lifecycle validation. Integration with the revised Chapter 4 ensures that documentation requirements and data management are harmonized across the digital value chain.
The introduction of Annex 22 represents a pivotal moment in the regulatory landscape for pharmaceutical manufacturing in Europe. This annex is the EU’s first dedicated framework addressing the use of Artificial Intelligence (AI) and machine learning in the production of active substances and medicinal products, responding to the rapid digital transformation now reshaping the industry.
Annex 22 sets out explicit requirements to ensure that any AI-based systems integrated into GMP-regulated environments are rigorously controlled and demonstrably trustworthy. It starts by mandating that manufacturers clearly define the intended use of any AI model deployed, ensuring its purpose is scientifically justified and risk-appropriate.
Quality risk management forms the backbone of Annex 22. Manufacturers must establish performance metrics tailored to the specific application and product risk profile of AI, and they are required to demonstrate the suitability and adequacy of all data used for model training, validation, and testing. Strong data governance principles apply: manufacturers need robust controls over data quality, traceability, and security throughout the AI system’s lifecycle.
The annex foresees a continuous oversight regime. This includes change control processes for AI models, ongoing monitoring of performance to detect drift or failures, and formally documented procedures for human intervention where necessary. The emphasis is on ensuring that, even as AI augments or automates manufacturing processes, human review and responsibility remain central for all quality- and safety-critical steps.
By introducing these requirements, Annex 22 aims to provide sufficient flexibility to enable innovation, while anchoring AI applications within a robust regulatory framework that safeguards product quality and patient safety at every stage. Together with the updates to Chapter 4 and Annex 11, Annex 22 gives companies clear, actionable expectations for responsibly harnessing digital innovation in the manufacturing environment.
Life Cycle Integration, Analytical Validation, and AI/ML Guidance
Across global regulators, a clear consensus has taken shape: validation must be seen as a continuous lifecycle process, not as a “check-the-box” activity. The latest WHO technical reports, the USP’s evolving chapters (notably <1058> and <1220>), and the harmonized ICH Q14 all signal a new age of ongoing qualification, continuous assurance, change management, and systematic performance verification. The scope of validation stretches from the design qualification stage through annual review and revalidation after every significant change.
A parallel wave of guidance for AI and machine learning is cresting. The EMA, FDA, MHRA, and WHO are now releasing coordinated documents addressing everything from transparent model architecture and dataset controls to rigorous “human-in-the-loop” safeguards for critical manufacturing decisions, including the new draft Annex 22. Data governance—traceability, security, and data quality—has never been under more scrutiny.
Regulatory Body
Document Title
Publication Date
Status
Key Focus Areas
EMA
Reflection Paper on the Use of Artificial Intelligence in the Medicinal Product Lifecycle
Oct-24
Final
Risk-based approach for AI/ML development, deployment, and performance monitoring across product lifecycle including manufacturing
EMA/HMA
Multi-annual AI Workplan 2023-2028
Dec-23
Final
Strategic framework for European medicines regulatory network to utilize AI while managing risks
EMA
Annex 22 Artificial Intelligence
Jul-25
Draft
Establishes requirements for the use of AI and machine learning in the manufacturing of active substances and medicinal products.
FDA
Considerations for the Use of AI to Support Regulatory Decision Making for Drug and Biological Products
Feb-25
Draft
Guidelines for using AI to generate information for regulatory submissions
FDA
Discussion Paper on AI in the Manufacture of Medicines
May-23
Published
Considerations for cloud applications, IoT data management, regulatory oversight of AI in manufacturing
FDA/Health Canada/MHRA
Good Machine Learning Practice for Medical Device Development Guiding Principles
Mar-25
Final
10 principles to inform development of Good Machine Learning Practice
WHO
Guidelines for AI Regulation in Health Care
Oct-23
Final
Six regulatory areas including transparency, risk management, data quality
MHRA
AI Regulatory Strategy
Apr-24
Final
Strategic approach based on safety, transparency, fairness, accountability, and contestability principles
EFPIA
Position Paper on Application of AI in a GMP Manufacturing Environment
Sep-24
Published
Industry position on using existing GMP framework to embrace AI/ML solutions
The Time is Now
The world of validation is no longer controlled by periodic updates or leisurely transitions. Change is the new baseline. Regulatory authorities have codified the digital, risk-based, and globally harmonized future—are your systems, people, and partners ready?