The recently released draft of ICH Q3E addresses a critical gap that has persisted in pharmaceutical regulation for over two decades. Since the FDA’s 1999 Container Closure Systems guidance and the EMA’s 2005 Plastic Immediate Packaging Materials guideline, the regulatory landscape for extractables and leachables has remained fragmented across regions and dosage forms. This fragmentation has created significant challenges for global pharmaceutical companies, leading to inconsistent approaches, variable interpretation of requirements, and substantial regulatory uncertainty that ultimately impacts patient access to medicines.
The ICH Q3E guideline emerges from recognition that modern pharmaceutical development increasingly relies on complex drug-device combinations, novel delivery systems, and sophisticated manufacturing technologies that transcend traditional regulatory boundaries. Biologics, cell and gene therapies, combination products, and single-use manufacturing systems have created E&L challenges that existing guidance documents were never designed to address. The guideline’s comprehensive scope encompasses chemical entities, biologics, biotechnological products, and drug-device combinations across all dosage forms, establishing a unified framework that reflects the reality of contemporary pharmaceutical manufacturing.
The harmonization achieved through ICH Q3E extends beyond mere procedural alignment to establish fundamental scientific principles that can be applied consistently regardless of geographical location or specific regulatory jurisdiction. This represents a significant evolution from the current patchwork of guidance documents, each with distinct requirements and safety thresholds that often conflict or create unnecessary redundancy in global development programs.
Comprehensive Risk Management Framework Integration
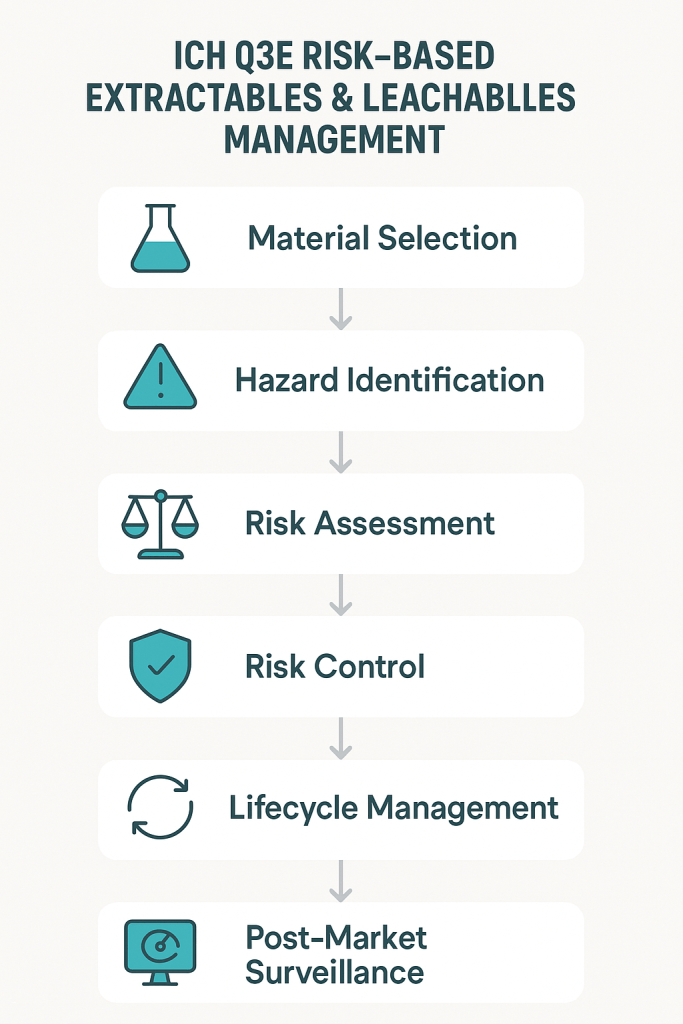
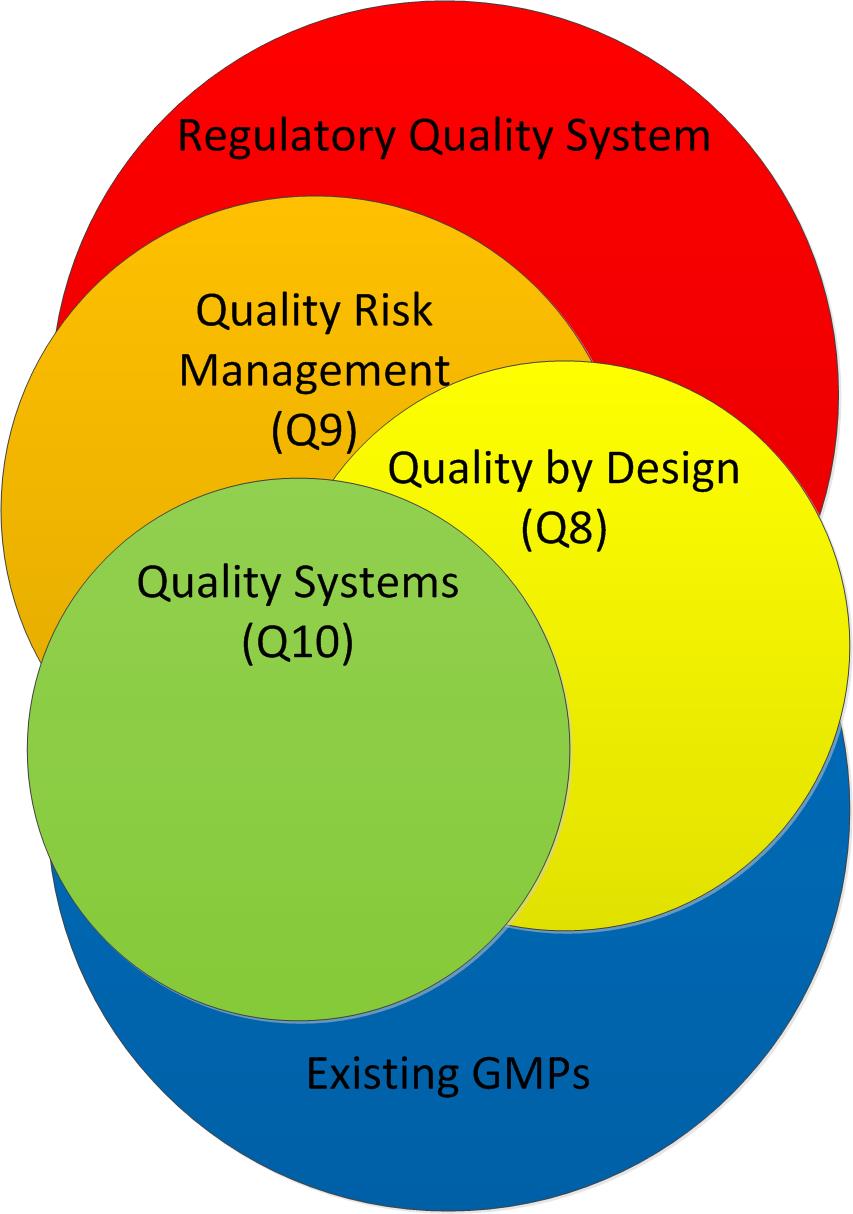
The most transformative aspect of ICH Q3E lies in its integration of comprehensive risk management principles derived from ICH Q9 throughout the entire E&L assessment process. This represents a fundamental departure from the prescriptive, one-size-fits-all approaches that have characterized previous guidance documents. The risk management framework encompasses four critical stages: hazard identification, risk assessment, risk control, and lifecycle management.
The hazard identification phase requires systematic evaluation of all materials of construction, manufacturing processes, and storage conditions that could contribute to extractables formation or leachables migration. This includes not only primary packaging components but also manufacturing equipment, single-use systems, filters, tubing, and any other materials that contact the drug substance or drug product during production, storage, or administration. The guideline recognizes that modern pharmaceutical manufacturing involves complex material interactions that require comprehensive evaluation beyond traditional container-closure system assessments.
Risk assessment under ICH Q3E employs a multi-dimensional approach that considers both the probability of extractables/leachables occurrence and the potential impact on product quality and patient safety. This assessment integrates factors such as contact time, temperature, pH, chemical compatibility, route of administration, patient population, and treatment duration. The framework explicitly acknowledges that risk varies significantly across different scenarios and requires tailored approaches rather than uniform requirements.
The risk control strategies outlined in ICH Q3E provide multiple pathways for managing identified risks, including material selection optimization, process parameter control, analytical monitoring, and specification limits. This flexibility enables pharmaceutical companies to develop cost-effective control strategies that are proportionate to the actual risks identified rather than applying maximum controls uniformly across all situations.
Lifecycle management ensures that E&L considerations remain integrated throughout product development, commercialization, and post-market surveillance. This includes provisions for managing material changes, process modifications, and the incorporation of new scientific knowledge as it becomes available. The lifecycle approach recognizes that E&L assessment is not a one-time activity but an ongoing process that must evolve with the product and available scientific understanding.
Safety Threshold Harmonization
ICH Q3E introduces a sophisticated threshold framework that harmonizes and extends the safety assessment principles developed through industry initiatives while addressing critical gaps in current approaches. The guideline establishes a risk-based threshold system that considers both mutagenic and non-mutagenic compounds while providing clear decision-making criteria for safety assessment.
For mutagenic compounds, ICH Q3E adopts a Threshold of Toxicological Concern (TTC) approach aligned with ICH M7 principles, establishing 1.5 μg/day as the default threshold for compounds with mutagenic potential. This represents harmonization with existing approaches while extending application to extractables and leachables that was previously addressed only through analogy or extrapolation.
For non-mutagenic compounds, the guideline introduces a tiered threshold system that considers route of administration, treatment duration, and patient population. The Safety Concern Threshold (SCT) varies based on these factors, with more conservative thresholds applied to high-risk scenarios such as parenteral administration or pediatric populations. This approach represents a significant advancement over current practice, which often applies uniform thresholds regardless of actual exposure scenarios or patient risk factors.
The Analytical Evaluation Threshold (AET) calculation methodology has been standardized and refined to provide consistent application across different analytical techniques and product configurations. The AET serves as the practical threshold for analytical identification and reporting, incorporating analytical uncertainty factors that ensure appropriate sensitivity for detecting compounds of potential safety concern.
The qualification threshold framework establishes clear decision points for when additional toxicological evaluation is required, reducing uncertainty and providing predictable pathways for safety assessment. Compounds below the SCT require no additional evaluation unless structural alerts are present, while compounds above the qualification threshold require comprehensive toxicological assessment using established methodologies.
Advanced Analytical Methodology Requirements
ICH Q3E establishes sophisticated analytical requirements that reflect advances in analytical chemistry and the increasing complexity of pharmaceutical products and manufacturing systems. The guideline requires fit-for-purpose analytical methods that are appropriately validated for their intended use, with particular emphasis on method capability to detect and quantify compounds at relevant safety thresholds.
The extraction study requirements have been standardized to ensure consistent generation of extractables profiles while allowing flexibility for product-specific optimization. The guideline establishes principles for solvent selection, extraction conditions, and extraction ratios that provide meaningful worst-case scenarios without introducing artifacts or irrelevant compounds. This standardization addresses a major source of variability in current practice, where different companies often use dramatically different extraction conditions that produce incomparable results.
Leachables assessment requirements emphasize the need for methods capable of detecting both known and unknown compounds in complex product matrices. The guideline recognizes the challenges associated with detecting low-level leachables in pharmaceutical formulations and provides guidance on method development strategies, including the use of placebo formulations, matrix subtraction approaches, and accelerated testing conditions that enhance detection capability.
The analytical uncertainty framework provides specific guidance on incorporating analytical variability into safety assessments, ensuring that measurement uncertainty does not compromise patient safety. This includes requirements for response factor databases, analytical uncertainty calculations, and the application of appropriate safety factors that account for analytical limitations.
Method validation requirements are tailored to the specific challenges of E&L analysis, including considerations for selectivity in complex matrices, detection limit requirements based on safety thresholds, and precision requirements that support reliable safety assessment. The guideline acknowledges that traditional pharmaceutical analytical validation approaches may not be directly applicable to E&L analysis and provides modified requirements that reflect the unique challenges of this application.
Material Science Integration and Innovation
ICH Q3E represents a significant advancement in the integration of material science principles into pharmaceutical quality systems. The guideline requires comprehensive material characterization that goes beyond simple compositional analysis to include understanding of manufacturing processes, potential degradation pathways, and interaction mechanisms that could lead to extractables formation.
The material selection guidance emphasizes proactive risk assessment during early development stages, enabling pharmaceutical companies to make informed material choices that minimize E&L risks rather than simply characterizing risks after materials have been selected. This approach aligns with Quality by Design principles and can significantly reduce development timelines and costs by avoiding late-stage material changes necessitated by unacceptable E&L profiles.
Single-use system assessment requirements reflect the increasing adoption of disposable manufacturing technologies in pharmaceutical production. The guideline provides specific frameworks for evaluating complex single-use assemblies that may contain multiple materials of construction and require additive risk assessment approaches. This addresses a critical gap in current guidance documents that were developed primarily for traditional reusable manufacturing equipment.
The guideline also addresses emerging materials and manufacturing technologies, including 3D-printed components, advanced polymer systems, and novel coating technologies. Provisions for evaluating innovative materials ensure that regulatory frameworks can accommodate technological advancement without compromising patient safety.
Comparison with Current Regulatory Frameworks
The transformative nature of ICH Q3E becomes evident when compared with existing regulatory approaches across different jurisdictions and application areas. The FDA’s 1999 Container Closure Systems guidance, while foundational, provides limited specific requirements and relies heavily on case-by-case assessment. This approach has led to significant variability in regulatory expectations and industry practice, creating uncertainty for both applicants and reviewers.
The EMA’s 2005 Plastic Immediate Packaging Materials guideline focuses specifically on plastic packaging materials and does not address the broader range of materials and applications covered by ICH Q3E. Additionally, the EMA guideline lacks specific safety thresholds, requiring product-specific risk assessment that can lead to inconsistent outcomes.
USP chapters <1663> and <1664> provide valuable technical guidance on extraction and leachables testing methodologies but do not establish safety thresholds or comprehensive risk assessment frameworks. These chapters serve as important technical references but require supplementation with safety assessment approaches from other sources.
The PQRI recommendations for orally inhaled and nasal drug products (OINDP) and parenteral and ophthalmic drug products (PODP) have provided industry-leading approaches to threshold-based safety assessment. However, these recommendations are limited to specific dosage forms and have not been formally adopted as regulatory requirements. ICH Q3E harmonizes and extends these approaches across all dosage forms while incorporating them into a formal regulatory framework.
Current European Pharmacopoeia requirements focus primarily on elemental extractables and do not address organic compounds comprehensively. The new EP chapter 2.4.35 on extractable elements represents an important advance but remains limited in scope compared to the comprehensive approach established by ICH Q3E.
ICH Q3E represents not merely an update or harmonization of existing approaches but a fundamental reconceptualization of E&L assessment that integrates the best elements of current practice while addressing critical gaps and inconsistencies.
Manufacturing Process Integration and Single-Use Systems
ICH Q3E places unprecedented emphasis on manufacturing process-related extractables and leachables, recognizing that modern pharmaceutical production increasingly relies on single-use systems, filters, tubing, and other disposable components that can contribute significantly to the overall E&L burden. This represents a major expansion from traditional container-closure system focus to encompass the entire manufacturing process.
The guideline establishes risk-based approaches for evaluating manufacturing equipment that consider factors such as contact time, process conditions, downstream processing steps, and the cumulative impact of multiple single-use components. This additive assessment approach acknowledges that even individually low-risk components can contribute to significant overall E&L levels when multiple components are used in series.
Single-use system assessment requirements address the complexity of modern bioprocessing equipment that may contain dozens of different materials of construction in a single assembly. The guideline provides frameworks for component-level assessment, assembly-level evaluation, and process-level integration that enable comprehensive risk assessment while maintaining practical feasibility.
The integration of manufacturing process E&L assessment with traditional container-closure system evaluation provides a holistic view of potential patient exposure that reflects the reality of modern pharmaceutical manufacturing. This comprehensive approach ensures that all sources of potential extractables and leachables are identified and appropriately controlled.
Biological Product Considerations and Specialized Applications
ICH Q3E provides specific considerations for biological products that reflect the unique challenges associated with protein stability, immunogenicity risk, and complex formulation requirements. Biological products often require specialized container-closure systems, delivery devices, and manufacturing processes that create distinct E&L challenges not adequately addressed by approaches developed for small molecule drugs.
The guideline addresses the potential for extractables and leachables to impact protein stability, aggregation, and biological activity through mechanisms that may not be captured by traditional chemical analytical approaches. This includes consideration of subvisible particle formation, protein adsorption, and catalytic degradation pathways that can be initiated by trace levels of extractables or leachables.
Immunogenicity considerations are explicitly addressed, recognizing that even very low levels of certain extractables or leachables could potentially trigger immune responses in sensitive patient populations. The guideline provides frameworks for assessing immunogenic risk that consider both the chemical nature of potential leachables and the clinical context of the biological product.
Cell and gene therapy applications receive special attention due to their unique manufacturing requirements, complex delivery systems, and often highly vulnerable patient populations. The guideline provides tailored approaches for these emerging therapeutic modalities that reflect their distinct risk profiles and manufacturing challenges.
Analytical Method Development and Validation Evolution
The analytical requirements established by ICH Q3E requires method capabilities that extend beyond traditional pharmaceutical analysis to encompass broad-spectrum unknown identification and quantification in complex matrices. This creates both challenges and opportunities for analytical laboratories and method development organizations.
Method development requirements emphasize systematic approaches to achieving required detection limits while maintaining selectivity in complex product matrices. The guideline provides specific guidance on extraction efficiency verification, matrix effect assessment, and the development of appropriate reference standards for quantification. These requirements ensure that analytical methods provide reliable data for safety assessment while maintaining practical feasibility.
Validation requirements are tailored to the unique challenges of E&L analysis, including considerations for compound identification confidence, quantification accuracy across diverse chemical structures, and method robustness across different product matrices. The guideline acknowledges that traditional pharmaceutical validation approaches may not be appropriate for E&L methods and provides modified requirements that reflect the specific challenges of this application.
The requirement for analytical uncertainty assessment and incorporation into safety evaluation represents a significant advancement in analytical quality assurance. Methods must not only provide accurate results but must also provide reliable estimates of measurement uncertainty that can be incorporated into risk assessment calculations.
Global Implementation Challenges and Opportunities
The implementation of ICH Q3E will require significant changes in pharmaceutical company practices, analytical capabilities, and regulatory review processes across all ICH regions. The comprehensive nature of the guideline means that virtually all pharmaceutical products will be impacted to some degree, creating both implementation challenges and opportunities for improved efficiency.
Training requirements will be substantial, as the guideline requires expertise in materials science, analytical chemistry, toxicology, and risk assessment that may not currently exist within all pharmaceutical organizations. The development of specialized E&L expertise will become increasingly important as companies seek to implement the guideline effectively.
Analytical infrastructure requirements may necessitate significant investments in instrumentation, method development capabilities, and reference standards. Smaller pharmaceutical companies may need to partner with specialized contract laboratories to access the required analytical capabilities.
Regulatory review processes will need to evolve to accommodate the risk-based approaches and comprehensive documentation requirements established by the guideline. Regulatory authorities will need to develop expertise in E&L assessment and establish consistent review practices across different therapeutic areas and product types.
The opportunities created by ICH Q3E implementation include improved regulatory predictability, reduced development timelines through early risk identification, and enhanced patient safety through more comprehensive E&L assessment. The harmonized approach should reduce the regulatory burden associated with multi-regional submissions while improving the overall quality of E&L assessments.
Future Evolution and Emerging Technologies
ICH Q3E has been designed with sufficient flexibility to accommodate emerging technologies and evolving scientific understanding. The risk-based framework can be adapted to new materials, manufacturing processes, and delivery systems as they are developed and implemented.
The guideline’s emphasis on scientific principles rather than prescriptive requirements enables adaptation to technological advances such as continuous manufacturing, advanced drug delivery systems, and personalized medicine approaches. This forward-looking design ensures that the guideline will remain relevant as pharmaceutical technology continues to evolve.
Provisions for incorporating new toxicological data and analytical methodologies ensure that the guideline can evolve with advancing scientific understanding. The lifecycle management approach enables updates and refinements based on accumulated experience and emerging scientific knowledge.
The integration with other ICH guidelines creates synergies that will facilitate future development of related guidance documents and ensure consistency across the broader ICH framework. This systematic approach to guideline development enhances the overall effectiveness of international pharmaceutical regulation.
Economic Impact and Industry Transformation
The implementation of ICH Q3E will have significant economic implications for the pharmaceutical industry, both in terms of implementation costs and long-term benefits. Initial implementation will require substantial investments in analytical capabilities, personnel training, and process modifications. However, the long-term benefits of harmonized requirements, improved regulatory predictability, and enhanced product quality are expected to provide significant value.
The harmonized approach should reduce the overall cost of global product development by eliminating duplicate testing requirements and reducing regulatory review timelines. Companies will be able to develop single global E&L strategies rather than maintaining multiple region-specific approaches.
Contract research organizations and analytical service providers will need to develop specialized capabilities to support pharmaceutical company implementation efforts. This will create new market opportunities while requiring significant investments in infrastructure and expertise.
The enhanced focus on risk-based assessment should enable more efficient allocation of resources to genuine safety concerns while reducing unnecessary testing and evaluation activities. This optimization of effort should improve overall industry efficiency while enhancing patient safety.
Patient Safety Enhancement and Risk Mitigation
The ultimate objective of ICH Q3E is enhanced patient safety through more comprehensive and scientifically rigorous assessment of extractables and leachables risks. The guideline achieves this objective through multiple mechanisms that address current gaps and limitations in E&L assessment practice.
The comprehensive material assessment requirements ensure that all potential sources of extractables and leachables are identified and evaluated. This includes not only traditional packaging materials but also manufacturing equipment, delivery device components, and any other materials that could contribute to patient exposure.
The harmonized safety threshold framework provides consistent and scientifically defensible criteria for safety assessment across all product types and administration routes. This eliminates the variability and uncertainty that can arise from inconsistent threshold application in current practice.
The risk-based approach enables appropriate allocation of assessment effort to genuine safety concerns while avoiding unnecessary evaluation of trivial risks. This optimization ensures that resources are focused on protecting patient safety rather than simply meeting regulatory requirements.
The lifecycle management requirements ensure that E&L considerations remain current throughout product development and commercialization. This ongoing attention to E&L issues helps identify and address emerging risks that might not be apparent during initial assessment.
Conclusion
ICH Q3E represents far more than an incremental improvement in extractables and leachables guidance; it establishes a new paradigm for pharmaceutical quality assurance that integrates materials science, analytical chemistry, toxicology, and risk management into a comprehensive framework that reflects the complexity of modern pharmaceutical development and manufacturing.
The guideline’s emphasis on scientific principles over prescriptive requirements creates a flexible framework that can accommodate the diverse and evolving landscape of pharmaceutical products while maintaining rigorous safety standards. This approach represents a significant maturation of regulatory science that moves beyond one-size-fits-all requirements to embrace risk-based, scientifically defensible assessment approaches.
The global harmonization achieved through ICH Q3E addresses one of the most significant challenges facing the pharmaceutical industry by providing consistent requirements and expectations across all major regulatory jurisdictions. This harmonization will facilitate more efficient global product development while enhancing patient safety through improved assessment practices.
The comprehensive scope of ICH Q3E ensures that extractables and leachables assessment evolves from a specialized concern for specific dosage forms to an integral component of pharmaceutical quality assurance across all products and therapeutic modalities. This integration reflects the reality that E&L considerations impact virtually all pharmaceutical products and must be systematically addressed throughout development and commercialization.
As the pharmaceutical industry prepares for ICH Q3E implementation, the focus must be on building the scientific expertise, analytical capabilities, and quality systems necessary to realize the guideline’s potential for enhancing patient safety while improving development efficiency. The successful implementation of ICH Q3E will mark a new era in pharmaceutical quality assurance that better serves patients, regulators, and the pharmaceutical industry through more rigorous, consistent, and scientifically defensible approaches to extractables and leachables assessment.
The transformation initiated by ICH Q3E extends beyond technical requirements to encompass fundamental changes in how pharmaceutical companies approach material selection, process design, analytical strategy, and risk management. This holistic transformation will ultimately deliver safer, higher-quality pharmaceutical products to patients worldwide while establishing a more efficient and predictable regulatory environment that facilitates innovation and global access to medicines.

{kind=link}