What the Warning Letter Reveals About Process Validation
The FDA’s inspection identified several violations that directly pertain to inadequate process validation. Process validation is essential for ensuring that drug manufacturing processes consistently produce products meeting their intended specifications. Here are the notable findings:
Failure to Validate Sterilization Processes:
The firm did not establish adequate controls to prevent microbiological contamination in drug products purporting to be sterile. Specifically, it relied on sterilization processes without monitoring pre-sterilization bioburden or maintaining appropriate environmental conditions.
The FDA emphasized that sterility testing alone is insufficient to assure product safety. It must be part of a broader validation strategy that includes pre-sterilization controls and environmental monitoring.
Inadequate Validation of Controlled-Release Dosage Forms:
The company failed to demonstrate that its controlled-release products conformed to specifications for active ingredient release rates. This lack of validation raises concerns about therapeutic efficacy and patient safety.
The response provided by the firm was deemed inadequate as it lacked retrospective assessments of marketed products and a detailed plan for corrective actions.
Insufficient Procedures for Production and Process Control:
The firm increased batch sizes without validating the impact on product quality and failed to include critical process parameters in batch records.
The FDA highlighted the importance of process qualification studies, which evaluate intra-batch variations and establish a state of control before commercial distribution.
Key Learnings for Pharmaceutical Manufacturers
The violations outlined in this warning letter provide valuable lessons for manufacturers aiming to maintain CGMP compliance:
Comprehensive Process Validation is Non-Negotiable
Process validation must encompass all stages of manufacturing, from raw materials to finished products. Manufacturers should:
Conduct rigorous qualification studies before scaling up production.
Validate sterilization processes, including pre-sterilization bioburden testing, environmental controls, and monitoring systems.
Sterility Testing Alone is Insufficient
Sterility testing should complement other preventive measures rather than serve as the sole assurance mechanism. Manufacturers must implement controls throughout the production lifecycle to minimize contamination risks.
Quality Control Units Must Exercise Oversight
The role of quality control units (QU) is pivotal in ensuring compliance across all operations, including oversight of contract testing laboratories and contract manufacturing organizations (CMOs). Failure to enforce proper testing protocols can lead to regulatory action.
Repeat Violations Signal Systemic Failures
The letter noted repeated violations from prior inspections in 2019 and 2021, indicating insufficient executive management oversight.
Through the skilled work of a very helpful FOIA officer at the FDA I have been reviewing the 2020 483 and EIR for the pre-approval inspection at the Sanofi Framingham, MA site that recently received a Warning Letter:
The 2020 pre-approval inspection (PAI) of Sanofi’s facility in Framingham, MA, uncovered critical deviations that exposed systemic weaknesses in contamination controls, equipment maintenance, and quality oversight. These deficiencies, documented in FDA Form 483 (FEI 1220423), violated 21 CFR 211 regulations and FDA Compliance Program 7346.832 requirements for PAIs. The facility’s failure to address these issues and to make systeatic changes over time (and perhaps backslide, but that is conjecture) contributed to subsequent regulatory actions, including a 2022 Form 483 and the 2024 FDA warning letter citing persistent CGMP violations. This analysis traces the 2020 findings to their regulatory origins, examines their operational consequences, and identifies lessons for PAI preparedness in high-risk API manufacturing.
Regulatory Foundations of Pre-Approval Inspections
The FDA’s PAI program operates under Compliance Program 7346.832, which mandates rigorous evaluation of facilities named in NDAs, ANDAs, or BLAs. Three pillars govern these inspections:
Commercial Manufacturing Readiness: PAIs assess whether facilities can reliably execute commercial-scale processes while maintaining CGMP compliance. This includes verification of validated equipment cleaning procedures, environmental monitoring systems, and preventive maintenance programs. The FDA prioritizes sites handling novel APIs, narrow therapeutic index drugs, or first-time applications—criteria met by Sanofi’s production of drug substances.
Application Conformance: Inspectors cross-validate submission data against actual operations, focusing on batch records, process parameters, and analytical methods. Discrepancies between filed documentation and observed practices constitute major compliance risks, particularly for facilities like Sanofi that utilize complex biologics manufacturing processes.
Data Integrity Assurance Per 21 CFR 211.194, PAIs include forensic reviews of raw data, equipment logs, and stability studies. The 2020 inspection identified multiple QC laboratory lapses at Sanofi that undermined data reliability—a red flag under FDA’s heightened focus on data governance in PAIs.
Facility Maintenance Deficiencies
Sterilization Equipment Contamination On September 2, 2020, FDA investigators documented (b)(4) residue on FB-2880-001 sterilization equipment and its transport cart—critical infrastructure for bioreactor probe sterilization. The absence of cleaning procedures or routine inspections violated 21 CFR 211.67(a), which mandates written equipment maintenance protocols. This lapse created cross-contamination risks for (b)(4) drug substances, directly contradicting the application’s sterility claims.
The unvalidated cleaning process for those chambers further breached 21 CFR 211.63, requiring equipment design that prevents adulteration. Historical data from 2008–2009 FDA inspections revealed similar sterilization issues at Allston facility, suggesting systemic quality control failures which suggests that these issues never were really dealt with systematically across all sites under the consent decree.
Environmental Control Breakdowns The August 26, 2020 finding of unsecured pre-filters in Downflow Booth —a critical area for raw material weighing—exposed multiple CGMP violations:
21 CFR 211.46(b): Failure to maintain HEPA filter integrity in controlled environments
FDA Aseptic Processing Guidance: Loose filters compromise ISO 5 unidirectional airflow
21 CFR 211.42(c): Inadequate facility design for preventing material contamination
Ceiling diffuser screens in Suite CNC space with unsecured fasteners exacerbated particulate contamination risks. The cumulative effect violated PAI Objective 1 by demonstrating poor facility control—a key factor in the 2024 warning letter’s citation of “unsuitable equipment for microbiologically controlled environments”.
Quality Control Laboratory Failures
Analytical Balance Non-Compliance The QC microbiology laboratory’s use of an unqualified balance breached multiple standards:
21 CFR 211.68(a): Lack of calibration for automated equipment
USP <41> Guidelines: Failure to establish minimum weigh limits
FDA Data Integrity Guidance (2018): Unguaranteed accuracy of microbiological test results
This deficiency directly impacted the reliability of bioburden testing data submitted in the application, contravening PAI Objective 3’s data authenticity requirements.
Delayed Logbook Reviews Three QC logbooks exceeded the review window specified in the site’s procedure:
Temperature logs for water baths
Dry state storage checklists
The delays violated 21 CFR 211.188(b)(11), which requires contemporaneous review of batch records. More critically, they reflected inadequate quality unit oversight—a recurring theme in Sanofi’s 2024 warning letter citing “lackluster quality control”.
And if they found 3 logbooks, chances are there were many more in an equal state.
Leak Investigations – A Leading Indicator
there are two pages in the EIR around leak deviation investigations, including the infamous bags, and in hindsight, I think this is an incredibly important inflection point from improvement that was missed.
The inspector took the time to evaluate quite a few deviations and overall control strategy for leaks and gave Sanofi a clean-bill of health. So we have to wonder if there was not enough problems to go deep enough to see a trend or if a sense of complacency allowed Sanofi to lower their guard around this critical aspect of single use, functionally closed systems.
The FDA’s July 2022 reinspection of Sanofi’s Framingham facility revealed persistent deficiencies despite corrective actions taken after the 2020 PAI. The inspection, conducted under Compliance Program 7356.002M, identified critical gaps in data governance and facility maintenance, resulting in a 2-item Form FDA 483 and an Official Action Indicated (OAI) classification – a significant escalation from the 2020 Voluntary Action Indicated (VAI) status.
Computerized System Control Failures
The FDA identified systemic weaknesses in data integrity controls for testers used to validate filter integrity during drug substance manufacturing. These testers generated electronic logs documenting failed and canceled tests that were never reviewed or documented in manufacturing records. For example:
On June 9, 2022, a filter underwent three consecutive tests for clarification operations: two failures and one cancellation due to operator error (audible “hissing” during testing). Only the final passing result was recorded in logbooks.
Between 2020–2022, operators canceled 14% of tests across testers without documented justification, violating 21 CFR 211.68(b) requirements for automated equipment review.
The firm had improperly classified these testers as “legacy electronic equipment,” bypassing mandatory audit trail reviews under their site procedure. I am not even sure what legacy electronic equipment means, but this failure contravened FDA’s Data Integrity Guidance (2018), which requires full traceability of GxP decisions.
Biological Safety Cabinet: Rust particles and brown residue contaminated interior surfaces used for drug substance handling in April 20223. The material was later identified as iron oxide from deteriorating cabinet components.
HVAC System Leaks: A pH probe in the water system leaked into grade-D areas, with standing water observed near active bioreactors3.
Structural Integrity Issues
Chipped epoxy floors in grade-C rooms created particulate generation risks during cell culture operations.
Improperly sloped flooring allowed pooling of rinse water adjacent to purification equipment.
These conditions violated 21 CFR 211.42(c), requiring facilities to prevent contamination through proper design, and demonstrated backsliding from 2020 corrective actions targeting environmental controls.
Regulatory Reckoning
These cultural failures crystallized in FDA’s 2024 citation of “systemic indifference to quality stewardship”. While some technological upgrades provided tactical fixes, the delayed recognition of cultural rot as root cause transformed manageable equipment issues into existential compliance threats—a cautionary tale for pharmaceutical manufacturers navigating dual challenges of technological modernization and workforce transition.
Conclusion: A Compliance Crisis Decade
The Sanofi case (2020–2024) exemplifies the consequences of treating PAIs as checklist exercises rather than opportunities for quality system maturation. The facility’s progression from 483 observations to OAI status and finally warning letter underscores three critical lessons:
Proactive Data Governance: Holitisic data overnance and data integrity, including audit trail reviews that encompass all GxP systems – legacy or modern.
Cultural Transformation: Quality metrics must drive executive incentives to prevent recurrent failures.
Manufacturers must adopt holistic systems integrating advanced analytics, robust knowledge management, and cultural accountability to avoid a costly regulatory debacle.
PAI Readiness Best Practices
Pre-Inspection Preparation
Gap Analysis Against CPGM 7346.832 Facilities should conduct mock inspections evaluating:
Conformance between batch records and application data
Completeness of method validation protocols
Environmental monitoring trend reports
Data Integrity Audits Forensic reviews of electronic records (e.g., HPLC chromatograms, equipment logs) using FDA’s “ALCOA+” criteria—ensuring data is Attributable, Legible, Contemporaneous, Original, and Accurate.
Facility Hardening Preventive maintenance programs for critical utilities:
Steam-in-place systems
HVAC airflow balances
Water for injection loops
Post-Approval Vigilance
The Sanofi case underscores the need for ongoing compliance monitoring post-PAI:
Quality Metrics Tracking: FDA-required metrics like lot rejection rates and CAPA effectiveness
Regulatory Intelligence: Monitoring emerging focus areas through FDA warning letters and guidance updates
Process Robustness Studies: Continued process verification per 21 CFR 211.110(a)
Reading Strukmyer LLC’s recent FDA Warning Letter, and reflecting back to last year’s Colgate-Palmolive/Tom’s of Maine, Inc. Warning Letter, has me thinking of common language In both warning letters where the FDA asks for “A comprehensive, independent assessment of the design and control of your firm’s manufacturing operations, with a detailed and thorough review of all microbiological hazards.”
It is hard to read that as anything else than a clarion call to use a HACCP.
If that isn’t a HACCP, I don’t know what is. Given the FDA’s rich history and connection to the tool, it is difficult to imagine them thinking of any other tool. Sure, I can invent about 7 other ways to do that, but why bother when there is a great tool, full of powerful uses, waiting to be used that the regulators pretty much have in their DNA.
The Evolution of HACCP in FDA Regulation: A Journey to Enhanced Food Safety
The Hazard Analysis and Critical Control Points (HACCP) system has a fascinating history that is deeply intertwined with FDA regulations. Initially developed in the 1960s by NASA, the Pillsbury Company, and the U.S. Army, HACCP was designed to ensure safe food for space missions. This pioneering collaboration aimed to prevent food safety issues by identifying and controlling critical points in food processing. The success of HACCP in space missions soon led to its application in commercial food production.
In the 1970s, Pillsbury applied HACCP to its commercial operations, driven by incidents such as the contamination of farina with glass. This prompted Pillsbury to adopt HACCP more widely across its production lines. A significant event in 1971 was a panel discussion at the National Conference on Food Protection, which led to the FDA’s involvement in promoting HACCP for food safety inspections. The FDA recognized the potential of HACCP to enhance food safety standards and began to integrate it into its regulatory framework.
As HACCP gained prominence as a food safety standard in the 1980s and 1990s, the National Advisory Committee on Microbiological Criteria for Foods (NACMCF) refined its principles. The committee added preliminary steps and solidified the seven core principles of HACCP, which include hazard analysis, critical control points identification, establishing critical limits, monitoring procedures, corrective actions, verification procedures, and record-keeping. This structured approach helped standardize HACCP implementation across different sectors of the food industry.
A major milestone in the history of HACCP was the implementation of the Pathogen Reduction/HACCP Systems rule by the USDA’s Food Safety and Inspection Service (FSIS) in 1996. This rule mandated HACCP in meat and poultry processing facilities, marking a significant shift towards preventive food safety measures. By the late 1990s, HACCP became a requirement for all food businesses, with some exceptions for smaller operations. This widespread adoption underscored the importance of proactive food safety management.
The Food Safety Modernization Act (FSMA) of 2011 further emphasized preventive controls, including HACCP, to enhance food safety across the industry. FSMA shifted the focus from responding to food safety issues to preventing them, aligning with the core principles of HACCP. Today, HACCP remains a cornerstone of food safety management globally, with ongoing training and certification programs available to ensure compliance with evolving regulations. The FDA continues to support HACCP as part of its broader efforts to protect public health through safe food production and processing practices. As the food industry continues to evolve, the principles of HACCP remain essential for maintaining high standards of food safety and quality.
Why is a HACCP Useful in Biotech Manufacturing
The HACCP seeks to map a process – the manufacturing process, one cleanroom, a series of interlinked cleanrooms, or the water system – and identifies hazards (a point of contamination) by understanding the personnel, material, waste, and other parts of the operational flow. These hazards are assessed at each step in the process for their likelihood and severity. Mitigations are taken to reduce the risk the hazard presents (“a contamination control point”). Where a risk cannot be adequately minimized (either in terms of its likelihood of occurrence, the severity of its nature, or both), this “contamination control point” should be subject to a form of detection so that the facility has an understanding of whether the microbial hazard was potentially present at a given time, for a given operation. In other words, the “critical control point” provides a reasoned area for selecting a monitoring location. For aseptic processing, for example, the target is elimination, even if this cannot be absolutely demonstrated.
The HACCP approach can easily be applied to pharmaceutical manufacturing where it proves very useful for microbial control. Although alternative risk tools exist, such as Failure Modes and Effects Analysis, the HACCP approach is better for microbial control.
HACCP provides a systematic approach to identifying and controlling potential hazards throughout the production process.
Step 1: Conduct a Hazard Analysis
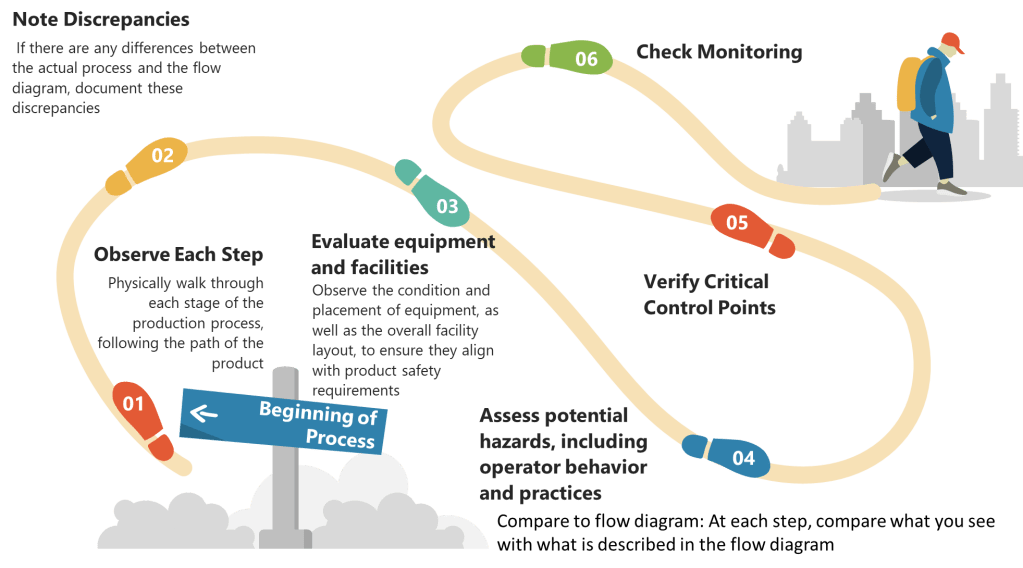
List All Process Steps: Begin by detailing every step involved in your biotech manufacturing process, from raw material sourcing to final product packaging. Make sure to walk down the process thoroughly.
Identify Potential Hazards: At each step, identify potential biological, chemical, and physical hazards. Biological hazards might include microbial contamination, while chemical hazards could involve chemical impurities or inappropriate reagents. Physical hazards might include particulates or inappropriate packaging materials.
Evaluate Severity and Likelihood: Assess the severity and likelihood of each identified hazard. This evaluation helps prioritize which hazards require immediate attention.
Determine Preventive Measures: Develop strategies to control significant hazards. This might involve adjusting process conditions, improving cleaning protocols, or enhancing monitoring systems.
Document Justifications: Record the rationale behind including or excluding hazards from your analysis. This documentation is essential for transparency and regulatory compliance.
Step 2: Determine Critical Control Points (CCPs)
Identify Control Points: Any step where biological, chemical, or physical factors can be controlled is considered a control point.
Determine CCPs: Use a decision tree to identify which control points are critical. A CCP is a step at which control can be applied and is essential to prevent or eliminate a hazard or reduce it to an acceptable level.
Establish Critical Limits: For each CCP, define the maximum or minimum values to which parameters must be controlled. These limits ensure that hazards are effectively managed.
Control Points
Critical Control Points
Process steps where a control measure (mitigation activity) is necessary to prevent the hazard from occurring
Process steps where both control and monitoring are necessary to assure product quality and patient safety
Are not necessarily critical control points (CCPs)
Are also control points
Determined from the risk associated with the hazard
Determined through a decision tree
Step 3: Establish Monitoring Procedures
Develop Monitoring Plans: Create detailed plans for monitoring each CCP. This includes specifying what to monitor, how often, and who is responsible.
Implement Monitoring Tools: Use appropriate tools and equipment to monitor CCPs effectively. This might include temperature sensors, microbial testing kits, or chemical analyzers.
Record Monitoring Data: Ensure that all monitoring data is accurately recorded and stored for future reference.
Step 4: Establish Corrective Actions
Define Corrective Actions: Develop procedures for when monitoring indicates that a CCP is not within its critical limits. These actions should restore control and prevent hazards.
Proceduralize: You are establishing alternative control strategies here so make sure they are appropriately verified and controlled by process/procedure in the quality system.
Train Staff: Ensure that all personnel understand and can implement corrective actions promptly.
Step 5: Establish Verification Procedures
Regular Audits: Conduct regular audits to verify that the HACCP system is functioning correctly. This includes reviewing monitoring data and observing process operations.
Validation Studies: Perform validation studies to confirm that CCPs are effective in controlling hazards.
Continuous Improvement: Use audit findings to improve the HACCP system over time.
Step 6: Establish Documentation and Record-Keeping
Maintain Detailed Records: Keep comprehensive records of all aspects of the HACCP system, including hazard analyses, CCPs, monitoring data, corrective actions, and verification activities.
Ensure Traceability: Use documentation to ensure traceability throughout the production process, facilitating quick responses to any safety issues.
Step 7: Implement and Review the HACCP Plan
Implement the Plan: Ensure that all personnel involved in biotech manufacturing understand and follow the HACCP plan.
Regular Review: Regularly review and update the HACCP plan to reflect changes in processes, new hazards, or lessons learned from audits and incidents.
I got a post on my RSS feed today from the FDA for a closeout letter to Safecor Health in response to the 2023 Warning Letter. Always happy to see an actual closeout letter.
The main takeaways from the FDA warning letter:
Inadequate Line Clearance and Packaging Controls:
Safecor failed to properly inspect packaging and labeling facilities before use, leading to potential mix-ups of drug products. This was evidenced by the presence of unrelated tablets and capsules during the packaging of a specific product.
The company has a history of product mix-ups, including instances where a vitamin was found in a drug meant to prevent organ transplant rejection and mislabeled blood clot prevention medication.
The firm lacked adequate procedures for cleaning and maintaining equipment, with unidentified residues found on supposedly clean equipment. This poses a risk of cross-contamination.
The company’s cleaning validation program was deemed inadequate, particularly in addressing worst-case scenarios.
Failure to Test Components:
Safecor did not adequately test incoming components, such as water used in drug manufacturing, for purity, strength, and quality.
The company relied on visual inspections without performing necessary chemical and microbiological tests.
Quality Control Unit Deficiencies:
The quality control unit failed to ensure compliance with CGMP regulations, including inadequate document control and data integrity issues.
Manufacturing records were not properly controlled, and corrections were made using correction fluid, raising concerns about data authenticity.
I get really confused on the differences between compounding pharmacies and outsourcing facilities. I’ve never worked at either, but see a lot of 483s and warning letters. So today I spent some snow day time doing some reading. I then wrote this up as a reminder to myself.
The Drug Quality and Security Act (DQSA) of 2013 introduced significant changes by distinguishing between compounding pharmacies under Section 503A and outsourcing facilities under Section 503B of the Federal Food, Drug, and Cosmetic Act (FDCA). This distinction is crucial for ensuring the safety and quality of compounded drugs.
Compounding Pharmacies (503A)
Definition and Purpose: Compounding pharmacies are licensed by state boards of pharmacy and primarily focus on creating customized medications for individual patients when commercially available drugs do not meet their needs. These pharmacies must adhere to standards set by the United States Pharmacopeia (USP), such as USP 797 for sterile compounding and USP 800 for hazardous drugs.
Regulatory Framework: Compounding pharmacies operate under the supervision of a licensed pharmacist and require a prescription for each compounded product. They are generally limited to small batches and are not allowed to engage in office-use compounding without a prescription.
Outsourcing Facilities (503B)
Definition and Purpose: Outsourcing facilities, on the other hand, are registered with the FDA and specialize in producing large batches of sterile drugs, often without the need for individual prescriptions. These facilities are designed to address drug shortages and provide complex or rarely compounded preparations to healthcare organizations.
Regulatory Framework: Unlike 503A pharmacies, 503B facilities must comply with FDA’s Current Good Manufacturing Practices (CGMP) to ensure the quality and safety of their products. They are subject to regular FDA inspections and must report on their compounded products.
Recent Regulatory Actions: The Case of ProRx, LLC
This research came about because I was reading a recent warning letter issued to ProRx, LLC, which basically stated they were failing to comply with CGMP regulations for 503B outsourcing facilities. The FDA identified serious deficiencies in sterile drug production practices, posing significant patient safety risks.
Implications for 503B Facilities
The warning letter to ProRx, LLC, serves as a reminder of the high regulatory bar set for 503B outsourcing facilities. Key implications include:
Enhanced Oversight: The FDA’s ability to inspect and enforce cGMP compliance means that 503B facilities must maintain meticulous quality control and production standards.
Patient Safety: The primary concern is ensuring that compounded drugs are safe for use. Facilities must address any deficiencies promptly to avoid recalls and protect patient health.
Partnerships and Supply Chain: The ability of 503B facilities to supply compounded drugs to healthcare organizations and pharmacies relies on their compliance with FDA regulations. Non-compliance can disrupt supply chains and impact patient access to necessary medications.
What I take away from my research is that 503B outsourcing facilities are GMP facilities, and are held to the same standard. Good to know as I evaluate their regulatory actions in the future. I think I’ve tended to dismiss them as not being in the same class of regulatory expectations.
Also, this is the second time this month where I really wonder what regulatory agencies fascination with pharmacists are in GMP facilities. Seems pretty clear to me that being a pharmacist is no indication of any capability around GMP activities.