There are two different ways that language is discussed and taught: descriptive grammar vs. prescriptive grammar.
Prescriptive grammar describes when people focus on talking about how a language should or ought to be used. Prescriptive grammar tells you how you should speak, and what type of language to avoid. This is commonly found in English classes where the aim is to teach people how to use language in a very particular (typically described as ‘proper’ or ‘correct’) way.
Descriptive grammar, on the other hand, focuses on describing the language as it is used, not saying how it should be used. For example, think about a prescriptive rule like Don’t split infinitives. A descriptive grammarian would see a sentence like “To boldly go where no man has gone before” and would try to describe how the mental grammar can cause that ordering of words, rather than saying that the surface form is faulty due to prescriptive rules (which would require the sentence “To go boldly where no man has gone before”). Linguistics takes this approach to language.
We have a similar thing in pharmaceutical regulations, often seen by how the FDA looks at certain issues and how the EMA and PIC/S looks at them.
For example, the FDA is proceeding with draft guidance on setting up inspection testing programs for detecting visible particles in injectable drugs is meant to address this issue from a good manufacturing practices (GMP) standpoint. This is mostly a descriptive approach, as it sets a lot of desirable outcomes but you few strong requirements for how to get there.
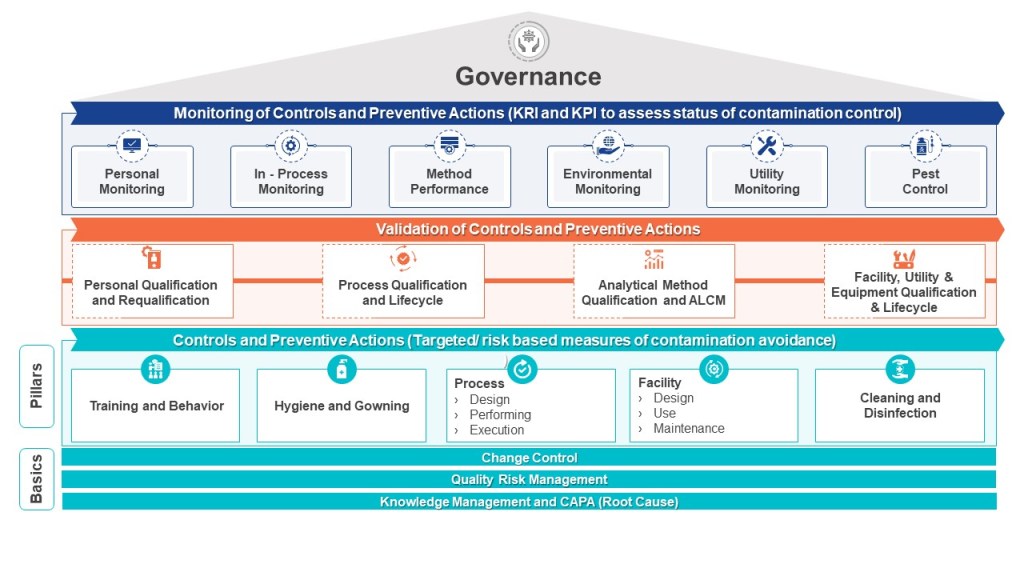
The EMA has a draft Annex 1 which lays out a pretty strong set of requirements for exactly how to perform contamination control, telling you exactly what to have and what it should look like.
The difference can be pretty evident when you hear the different regulators discuss their approaches. I’ve certainly heard more than one present or former FDA regulator say that adopting Annex 1 isn’t necessary because the GMPs already have the requirements built in.
You see a similar approach when it comes to QPs or the GCPs.
The prescriptive versus descriptive difference even comes up during inspections. Most people will talk about how the FDA focuses on artifacts and the EMA goes deep on the process.
A similar divide can happen in your quality system where you see different approaches (often a hodge-podge) between controlling the bad and promoting the good.
Being a pragmatist I often see benefits in both approaches (the same way I find value navigating between FDA and EMA approaches). The key thing is being deliberate about it.