As October rolls around I am focusing on 3 things: finalizing a budget; organization design and talent management; and a 2025 metrics plan. One can expect those three things to be the focus of a lot of my blog posts in October.
Go and read my post on Metrics plans. Like many aspects of a quality management system we don’t spend nearly enough time planning for metrics.
So over the next month I’m going to develop the strategy for a metrics plan to ensure the optimal performance, safety, and compliance of our biotech manufacturing facility, with a focus on:
Facility and utility systems efficiency
Equipment reliability and performance
Effective commissioning, qualification, and validation processes
Robust quality risk management
Stringent contamination control measures
Following the recommended structure of a metrics plan, here is the plan:
Rationale and Desired Outcomes
Implementing this metrics plan will enable us to:
Improve overall facility performance and product quality
Reduce downtime and maintenance costs
Ensure regulatory compliance
Minimize contamination risks
Optimize resource allocation
Metrics Framework
Our metrics framework will be based on the following key areas:
Facility and Utility Systems
Equipment Performance
Commissioning, Qualification, and Validation (CQV)
Quality Risk Management (QRM)
Contamination Control
Success Criteria
Success will be measured by:
Reduction in facility downtime
Improved equipment reliability
Faster CQV processes
Decreased number of quality incidents
Reduced contamination events
Implementation Plan
Steps, Timelines & Milestones
Develop detailed metrics for each key area (Month 1)
Implement data collection systems (Month 2)
Train personnel on metrics collection and analysis (Month 3)
Begin data collection and initial analysis (Month 4)
Review and refine metrics (Month 9)
Full implementation and ongoing analysis (Month 12 onwards)
This plan gets me ready to evaluate these metrics as part of governance in January of next year.
In October I will breakdown some metrics, explaining them and provide the rationale, and demonstrate how to collect. I’ll be striving to break these metrics into key performance indicators (KPI), key behavior indicators (KBI) and key risk indicators (KRI).
It is crucial for a Marketing Authorization Holder (MAH) to review and approve changes made by a Contract Development and Manufacturing Organization (CDMO) for several important reasons:
Regulatory Compliance
The Market Authorization Holder (MAH) – or the sponsor for pre-commercial GMP manufacturing – bears the primary responsibility for ensuring compliance with the marketing authorization and regulatory requirements throughout the product’s lifecycle. By reviewing and approving CDMO changes, the MAH can:
Ensure changes align with the approved marketing authorization
Verify that any variations to the marketing authorization are properly submitted to regulatory authorities
Maintain oversight of post-approval change management as required by regulations
Before I go any further on the topic I want you to go and read my post Classification of Changes for GMP/GDP. This post will build on that discussion.
I think it is better for the CDMO to put a lot of thought into this, and the MAH (the client) to evaluate and adapt. For all but the big players, the volume is going to be on the CDMO’s side. But if you are the client and your CDMO hasn’t taken this into account to the appropriate degree, you need to ensure appropriate steps taken. As such the rest of this post will be written from the CDMO’s side, but the same principles apply to the MAH (and should be included in the audit program).
Remember we have three goals:
Fulfill our contractual responsibilities
Help the MAH maintain appropriate control as the product owner
Ensure alignment between both parties on change implementation
The critical requirement here is ensuring the right changes get to the right client so they can be filled the right way. Returning to basics, we are approaching changes as:
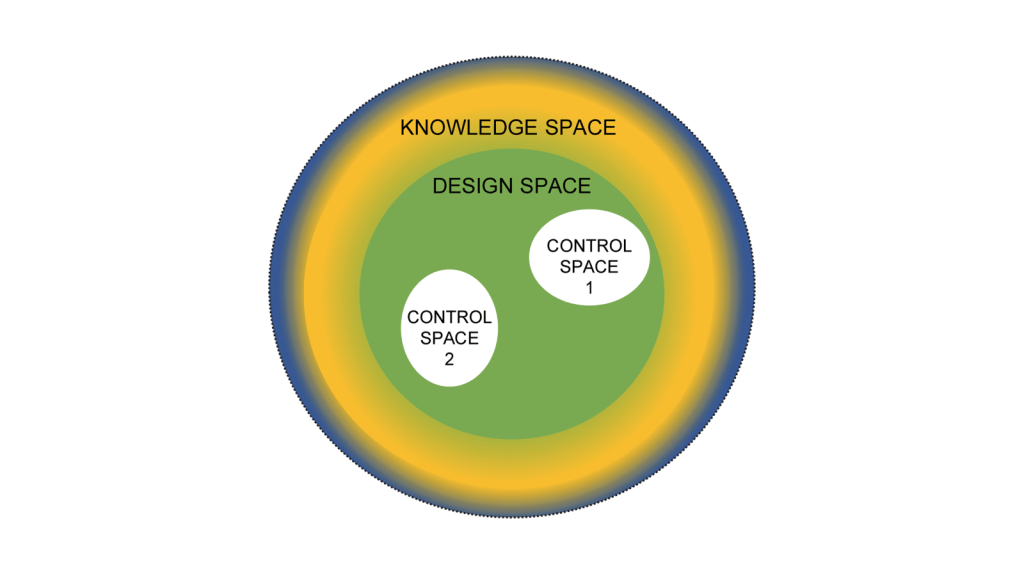
Now it’s easy to apply this to product. Create and/or receive the design space and the control space. Everything that falls into a non-established condition does not get reported to the client at time of execution. If it is “Do and Report” is is in the APQR. If it is “Do and Record” they can see it during the audit.
Where a lot of CDMOs trip up here are facility and quality system changes. My recommendation here is the same, define a design space based on the CMC section of the Common Technical Document which basically boils down to:
The CMC (Chemistry, Manufacturing, and Controls) section of a regulatory dossier typically includes the following key facility-related information:
Manufacturing Facilities
Names and addresses of all manufacturing, testing, and storage facilities involved in production
Description of the manufacturing operations performed at each site
Floor plans and layouts of production areas
Details on utilities and support systems (HVAC, water, gases, etc.)
Information on facility design features for contamination control and product protection
Equipment
List of major production and laboratory equipment
Equipment specifications and capacities
Cleaning and maintenance procedures for equipment
Environmental Controls
Description of clean room classifications and environmental monitoring programs
Air handling systems and controls
Water systems (purified water, water for injection) and controls
Material Flow
Personnel and material flow diagrams
Segregation of operations to prevent cross-contamination
Quality Control Laboratories
Description of QC lab facilities and equipment
Environmental controls in QC labs
Storage Areas
Description of storage facilities for raw materials, intermediates, and finished products
Storage conditions and controls (temperature, humidity, etc.)
There is a whole lot of wiggle room here in things that fall into “Do and Record.” By building this into your change control system you can delineate what goes to to the client and what doesn’t. I recommend sitting down with this list and deciding what types of changes fall into “Tell and Do” – what you ask permission from clients before doing; “Do and Report” – what goes in the APQR; and, “Do and Record” – what the client sees when they audit.
You know have good rules on what changes go to a client for prior approval and which ones do not. This gets codified in two places: the change control process and the quality/technical agreement.
Some other things to build into your change control process:
Documenting when a client requests a change, the reason and the impact on the platform. Remember you have other clients, and more and more CDMO’s are offering a platform, so there needs to be appropriate review and endorsement.
Think through how changes to facility (and other platform elements) are communicated and gated for multiple clients. Have a mechanism to manage client specific activities and to track first-product impacted for multiple products.
Have clear timelines and expectations on change communication and approval with the client in the quality/technical agreement. Hold each other accountable.
Have contingency plans. There will always be that one client who will be in shortage if you make that urgent change just when you want/need to.
Have a method for evaluating requested changes to the change plan by clients and making decisions around it. There will be that one client who doesn’t agree or wants something weird that disagrees with what all the other clients want.
Have rules in place to manage changes inactive for long periods or extensions specific for those changes that rise to client approval. These will have a different flow than internal changes.
I’ve used a bit of commercial headspace for this post, relying on the APQR. For clinical processes, product tends to fall into campaign-mindset, so “Do and Report” ends up being more a clinical campaign change report than an APQR.
Multiple studies have found that when approximately 25% of a population adopts a new behavior or belief, it can trigger a rapid shift toward widespread adoption. This suggests that change initiatives in organizations may gain critical momentum once about a quarter of employees get on board. The concept of a tipping point is really important in the development of a quality culture.
Factors Influencing Tipping Points
Several factors can affect where the tipping point occurs:
Strength of existing norms: More entrenched behaviors require larger minority groups to spark change.
Social costs: Higher penalties for non-conformity make change more difficult.
Visibility: Changes that are more observable spread more easily.
Incentives: Financial or other rewards for maintaining status quo can impede change.
Implications for Organizational Change
Based on this research, some key takeaways for driving change in organizations include:
Focus on early adopters: Concentrate efforts on getting 25-30% of employees to embrace the change initially.
Increase visibility: Make adoption of new behaviors highly visible to accelerate social contagion.
Reduce barriers: Minimize social or financial costs for early adopters of change.
Persistence is key: Change agents should persist even if initial efforts seem unsuccessful – they may be close to the tipping point.
The Role of Leadership
Leaders play a crucial role in engineering environments conducive to change. Given that they control key levers (incentives and social costs especially) not having leaders on board is devastating. Leaders need to:
Creating common understanding of benefits
Encouraging and supporting “change champions”
Aligning incentives with desired new behaviors
Facilitating rapid information flow about adoption
A recent FDA Warning Letter really drove home a good point about the perils of ‘retrospective validation’ and how that normally doesn’t mean what folks want it to mean.
“In lieu of process validation studies, you attempted to retrospectively review past batches without scientifically establishing blend uniformity and other critical process performance indicators. You do not commit to conduct further process performance qualification studies that scientifically establish the ability of your manufacturing process to consistently yield finished products that meet their quality attributes.”
The FDA’s response here is important for three truths:
Validation needs to be done against critical quality attributes and critical process parameters to scientifically establish that the manufacturing process is consistent.
Batch data on its own is rather useless.
Validation is a continuous exercise, it is not once-and-done (or rather in most people’s view thrice-and-done).
I don’t think the current GMPs really allow the concept of retrospective validation as most people want it to mean (including the recipient of that warning letter). It’s probably a term we should go into the big box of Nope.
AI generated art
Retrospective validation as most people mean it is a type of process validation that involves evaluating historical data and records to demonstrate that an existing process consistently produces products meeting predetermined specifications. As an approach retrospective validation involves evaluating historical data and records to demonstrate that an existing process consistently produces products meeting predetermined specifications.
The problem here is that this really just tells you what you were already hoping was true.
Retrospective validation has some major flaws:
Limited control over data quality and completeness: Since retrospective validation relies on historical data, there may be gaps or inconsistencies in the available information. The data may not have been collected with validation in mind, leading to missing critical parameters or measurements. It rather throws out most of the principles of science.
Potential bias in existing data: Historical data may be biased or incomplete, as it was not collected specifically for validation purposes. This can make it difficult to draw reliable conclusions about process performance and consistency.
Difficulty in identifying and addressing hidden flaws: Since the process has been in use for some time, there may be hidden flaws or issues that have not been identified or challenged. These could potentially lead to non-conforming products or hazardous operating conditions.
Difficulty in recreating original process conditions: It may be challenging to accurately recreate or understand the original process conditions under which the historical data was generated, potentially limiting the validity of conclusions drawn from the data.
What is truly called for is to perform concurrent validation.
The biotech industry is experiencing a significant transformation in validation processes, driven by rapid technological advancements, evolving regulatory standards, and the development of novel therapies.
The 2024 State of Validation report, authored by Jonathan Kay and funded by Kneat, provides a overview of trends and challenges in the validation industry. Here are some of the key findings:
Compliance and efficiency are top priorities: Creating process efficiencies and ensuring audit readiness have become the primary goals for validation programs.
Compliance burden emerged as the top validation challenge in 2024, replacing shortage of human resources which was the top concern in 2022-2023
Digital transformation is accelerating: 83% of respondents are either using or planning to adopt digital validation systems. The top benefits include improved data integrity, continuous audit readiness, and global standardization.
79% of those using digital validation rely on third-party software providers
Does this mean that 21% of respondents are in companies that have created their own bespoke systems? Or is something else going on there
63% reported that ROI from digital validation systems met or exceeded expectations
Artificial intelligence and machine learning are on the rise: 70% of respondents believe AI and ML will play a pivotal role in the future of validation.
Remote audits are becoming more common: 75% of organizations conducted at least some remote regulatory audits in the past year.
Challenges persist: The industry faces ongoing challenges in balancing costs, attracting talent, and keeping pace with technological advancements.
61% reported an increase in validation workload over the past 12 months
Industry 4.0 adoption is growing: 60% of organizations are in the early stages or actively implementing Industry/Pharma 4.0 technologies.
Digital Transformation:
As highlighted in the 2024 State of Validation report and my previous blog post on “Challenges in Validation,” several key trends and challenges are shaping the future of validation in biotech:
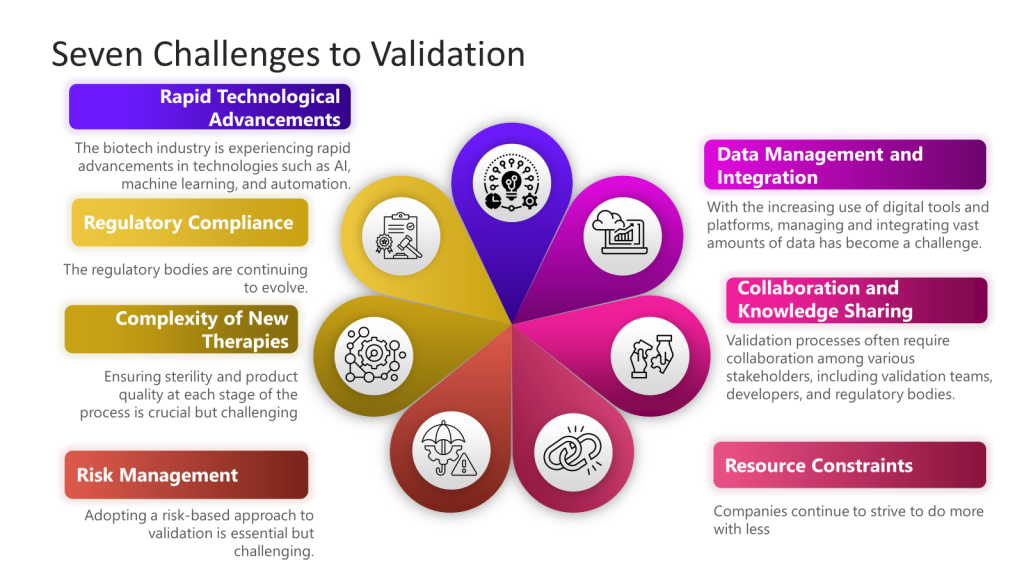
Technological Integration: The integration of AI, machine learning, and automation into validation processes presents both opportunities and challenges. While these technologies offer the potential for increased efficiency and accuracy, they also require new validation frameworks and methodologies.
Regulatory Compliance: Keeping pace with evolving regulatory standards remains a significant challenge. Regulatory bodies are continuously updating guidelines to address technological advancements, requiring companies to stay vigilant and adaptable.
Data Management and Integration: With the increasing use of digital tools and platforms, managing and integrating vast amounts of data has become a critical challenge. The industry is moving towards more robust data analytics and machine learning tools to handle this data efficiently.
Resource Constraints: Particularly for smaller biotech companies, resource limitations in funding, personnel, and expertise can hinder the implementation of advanced validation techniques.
Risk Management: Adopting a risk-based approach to validation is essential but challenging. Companies must develop effective strategies to identify and mitigate risks throughout the product lifecycle.
Collaboration and Knowledge Sharing: Ensuring effective communication and data sharing among various stakeholders is crucial for streamlining validation efforts and aligning goals.
Digital Transformation: The industry is witnessing a shift from traditional, paper-heavy validation methods to more dynamic, data-driven, and digitalized processes. This transformation promises enhanced efficiency, compliance, and collaboration.
Workforce Development: We are a heavily experience driven field. With 38% of validation professionals having 16 or more years of experience, there’s a critical need for knowledge transfer and training to equip newer entrants with necessary skills.
Adoption of Computer Software Assurance (CSA): The industry is gradually embracing CSA processes, driven by recent FDA guidance, though there’s still considerable room for further adoption. I always find this showing up in surveys to be disappointing, as CSA is a racket, as it basically is already existing validation principles. But consultants got to consult.
Focus on Efficiency and Audit Readiness: Creating process efficiencies and ensuring audit readiness have emerged as top priorities for validation programs.
As the validation landscape continues to evolve, it’s crucial for biotech companies to embrace these changes proactively. By leveraging new technologies, fostering collaboration, and focusing on continuous improvement, the industry can overcome these challenges and drive innovation in validation processes.
The future of validation in biotech lies in striking a balance between technological advancement and regulatory compliance, all while maintaining a focus on product quality and patient safety. As we move forward, it’s clear that the validation field will continue to be dynamic and exciting, offering numerous opportunities for innovation and growth.