The environment for commissioning, qualification, and validation (CQV) professionals remains defined by persistent challenges. Rapid technological advancements—most notably in artificial intelligence, machine learning, and automation—are constantly reshaping the expectations for validation. Compliance requirements are in frequent flux as agencies modernize guidance, while the complexity of novel biologics and therapies demands ever-higher standards of sterility, traceability, and process control. The shift towards digital systems has introduced significant hurdles in data management and integration, often stretching already limited resources. At the same time, organizations are expected to fully embrace risk-based, science-first approaches, which require new methodologies and skills. Finally, true validation now hinges on effective collaboration and knowledge-sharing among increasingly cross-functional and global teams.
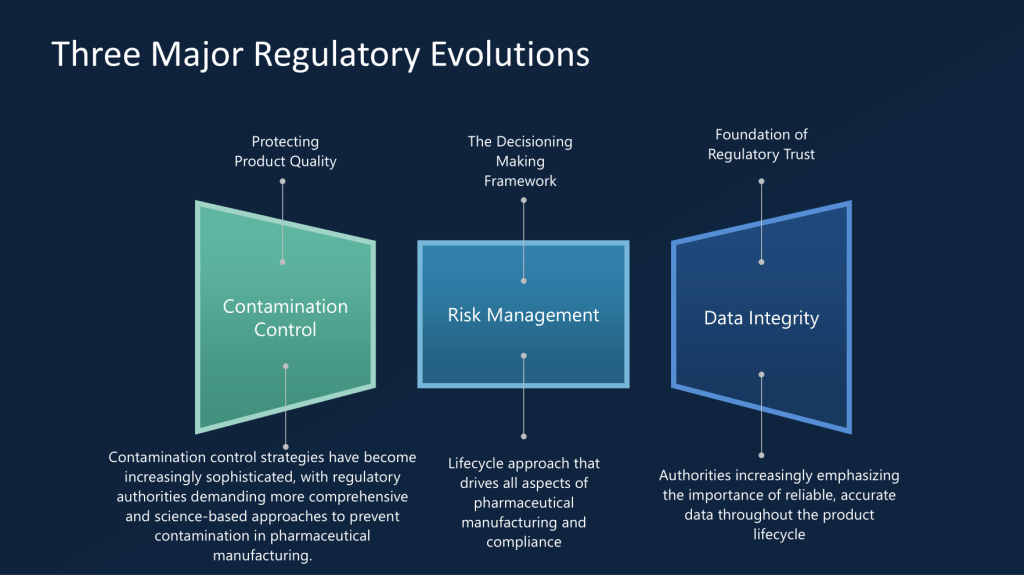
Overlaying these challenges, three major regulatory paradigm shifts are transforming the expectations around risk management, contamination control, and data integrity. Data integrity in particular has become an international touchpoint. Since the landmark PIC/S guidance in 2021 and matching World Health Organization updates, agencies have made it clear that trustworthy, accurate, and defendable data—whether paper-based or digital—are the foundation of regulatory confidence. Comprehensive data governance, end-to-end traceability, and robust documentation are now all non-negotiable.
Contamination control is experiencing its own revolution. The August 2023 overhaul of EU GMP Annex 1 set a new benchmark for sterile manufacturing. The core concept, the Contamination Control Strategy (CCS), formalizes expectations: every manufacturer must systematically identify, map, and control contamination risks across the entire product lifecycle. From supply chain vigilance to environmental monitoring, regulators are pushing for a proactive, science-driven, and holistic approach, far beyond previous practices that too often relied on reactive measures. We this reflected in recent USP drafts as well.
Quality risk management (QRM) also has a new regulatory backbone. The ICH Q9(R1) revision, finalized in 2023, addresses long-standing shortcomings—particularly subjectivity and lack of consistency—in how risks are identified and managed. The European Medicines Agency’s ongoing revision of EudraLex Chapter 1, now aiming for finalization in 2026, will further require organizations to embed preventative, science-based risk management within globalized and complex supply chain operations. Modern products and supply webs simply cannot be managed with last-generation compliance thinking.
The EU Digital Modernization: Chapter 4, Annex 11, and Annex 22
With the rapid digitalization of pharma, the European Union has embarked on an ambitious modernization of its GMP framework. At the heart of these changes are the upcoming revisions to Chapter 4 (Documentation), Annex 11 (Computerised Systems), and the anticipated implementation of Annex 22 (Artificial Intelligence).
Chapter 4—Documentation is being thoroughly updated in parallel with Annex 11. The current chapter, which governs all aspects of documentation in GMP environments, was last revised in 2011. Its modernization is a direct response to the prevalence of digital tools—electronic records, digital signatures, and interconnected documentation systems. The revised Chapter 4 is expected to provide much clearer requirements for the management, review, retention, and security of both paper and electronic records, ensuring that information flows align seamlessly with the increasingly digital processes described in Annex 11. Together, these updates will enable companies to phase out paper where possible, provided electronic systems are validated, auditable, and secure.
Annex 11—Computerised Systems will see its most significant overhaul since the dawn of digital pharma. The new guidance, scheduled for publication and adoption in 2026, directly addresses areas that the previous version left insufficiently covered. The scope now embraces the tectonic shift toward AI, machine learning, cloud-based services, agile project management, and advanced digital workflows. For instance, close attention is being paid to the robustness of electronic signatures, demanding multi-factor authentication, time-zoned audit trails, and explicit provisions for non-repudiation. Hybrid (wet-ink/digital) records will only be acceptable if they can demonstrate tamper-evidence via hashes or equivalent mechanisms. Especially significant is the regulation of “open systems” such as SaaS and cloud platforms. Here, organizations can no longer rely on traditional username/password models; instead, compliance with standards like eIDAS for trusted digital providers is expected, with more of the technical compliance burden shifting onto certified digital partners.
The new Annex 11 also calls for enhanced technical controls throughout computerized systems, proportional risk management protocols for new technologies, and a far greater emphasis on continuous supplier oversight and lifecycle validation. Integration with the revised Chapter 4 ensures that documentation requirements and data management are harmonized across the digital value chain.
The introduction of Annex 22 represents a pivotal moment in the regulatory landscape for pharmaceutical manufacturing in Europe. This annex is the EU’s first dedicated framework addressing the use of Artificial Intelligence (AI) and machine learning in the production of active substances and medicinal products, responding to the rapid digital transformation now reshaping the industry.
Annex 22 sets out explicit requirements to ensure that any AI-based systems integrated into GMP-regulated environments are rigorously controlled and demonstrably trustworthy. It starts by mandating that manufacturers clearly define the intended use of any AI model deployed, ensuring its purpose is scientifically justified and risk-appropriate.
Quality risk management forms the backbone of Annex 22. Manufacturers must establish performance metrics tailored to the specific application and product risk profile of AI, and they are required to demonstrate the suitability and adequacy of all data used for model training, validation, and testing. Strong data governance principles apply: manufacturers need robust controls over data quality, traceability, and security throughout the AI system’s lifecycle.
The annex foresees a continuous oversight regime. This includes change control processes for AI models, ongoing monitoring of performance to detect drift or failures, and formally documented procedures for human intervention where necessary. The emphasis is on ensuring that, even as AI augments or automates manufacturing processes, human review and responsibility remain central for all quality- and safety-critical steps.
By introducing these requirements, Annex 22 aims to provide sufficient flexibility to enable innovation, while anchoring AI applications within a robust regulatory framework that safeguards product quality and patient safety at every stage. Together with the updates to Chapter 4 and Annex 11, Annex 22 gives companies clear, actionable expectations for responsibly harnessing digital innovation in the manufacturing environment.
Life Cycle Integration, Analytical Validation, and AI/ML Guidance
Across global regulators, a clear consensus has taken shape: validation must be seen as a continuous lifecycle process, not as a “check-the-box” activity. The latest WHO technical reports, the USP’s evolving chapters (notably <1058> and <1220>), and the harmonized ICH Q14 all signal a new age of ongoing qualification, continuous assurance, change management, and systematic performance verification. The scope of validation stretches from the design qualification stage through annual review and revalidation after every significant change.
A parallel wave of guidance for AI and machine learning is cresting. The EMA, FDA, MHRA, and WHO are now releasing coordinated documents addressing everything from transparent model architecture and dataset controls to rigorous “human-in-the-loop” safeguards for critical manufacturing decisions, including the new draft Annex 22. Data governance—traceability, security, and data quality—has never been under more scrutiny.
Regulatory Body
Document Title
Publication Date
Status
Key Focus Areas
EMA
Reflection Paper on the Use of Artificial Intelligence in the Medicinal Product Lifecycle
Oct-24
Final
Risk-based approach for AI/ML development, deployment, and performance monitoring across product lifecycle including manufacturing
EMA/HMA
Multi-annual AI Workplan 2023-2028
Dec-23
Final
Strategic framework for European medicines regulatory network to utilize AI while managing risks
EMA
Annex 22 Artificial Intelligence
Jul-25
Draft
Establishes requirements for the use of AI and machine learning in the manufacturing of active substances and medicinal products.
FDA
Considerations for the Use of AI to Support Regulatory Decision Making for Drug and Biological Products
Feb-25
Draft
Guidelines for using AI to generate information for regulatory submissions
FDA
Discussion Paper on AI in the Manufacture of Medicines
May-23
Published
Considerations for cloud applications, IoT data management, regulatory oversight of AI in manufacturing
FDA/Health Canada/MHRA
Good Machine Learning Practice for Medical Device Development Guiding Principles
Mar-25
Final
10 principles to inform development of Good Machine Learning Practice
WHO
Guidelines for AI Regulation in Health Care
Oct-23
Final
Six regulatory areas including transparency, risk management, data quality
MHRA
AI Regulatory Strategy
Apr-24
Final
Strategic approach based on safety, transparency, fairness, accountability, and contestability principles
EFPIA
Position Paper on Application of AI in a GMP Manufacturing Environment
Sep-24
Published
Industry position on using existing GMP framework to embrace AI/ML solutions
The Time is Now
The world of validation is no longer controlled by periodic updates or leisurely transitions. Change is the new baseline. Regulatory authorities have codified the digital, risk-based, and globally harmonized future—are your systems, people, and partners ready?
Let’s not sugarcoat it: if you’re still allowing passwords like “Quality2025!” or “GMPpassword!” anywhere in your regulated workflow, you’re inviting trouble. The era of security theater is over. Modern cyberattacks and regulatory requirements—from NIST to EU GMP Annex 11—demand far more than adding an exclamation point to a dictionary word. It’s time to understand not just why dictionary words are dangerous, but how smart password strategy (including password managers) is now a fundamental part of data integrity and compliance.
Let’s start with why dictionary words are pure liability. Attackers don’t waste resources guessing random character strings—they leverage enormous “dictionary lists” sourced from real-world breaches, wordlists, and common phrases. Tools like Hashcat or John the Ripper process billions of guesses—including every English word and thousands of easy permutations—faster than you can blink.
This means that passwords like “Laboratory2025” or “Pharma@123” fall within minutes (or seconds) of an attack. Even a special character or a capital letter doesn’t save you, because password-cracking tools automatically try those combinations.
The Verizon Data Breach Investigations Report has put it plainly: dictionary attacks and credential stuffing remain some of the top causes for data compromise. If a GxP system accepts any plain-language word, it’s a red flag for any inspection—and a massive technical vulnerability.
What the Latest NIST Guidance Says
The definitive voice for password policy, the National Institute of Standards and Technology (NIST), made a seismic shift with Special Publication 800-63B (“Digital Identity Guidelines: Authentication and Lifecycle Management”). The relevant part:
“Verifiers SHALL compare…” NIST 800-63B Section 5.1.1.2 requires your system to check a new password against lists of known bad, common, or compromised passwords—including dictionary words. If it pops up anywhere, it’s out.
But NIST also dispelled the notion that pure complexity (“$” instead of “S”, “0” instead of “o”) provides security. Their new mantra is:
No dictionary words
No user IDs, product names, or predictable info
No passwords ever found in a breach
BUT: do make them long, unique, and easy to use with a password manager
Dictionary Words vs. Passphrases: Not All Words Are Bad—But Phrases Must Be Random
Many people hear “no dictionary words” and assume they must abandon human language. Not so! NIST recommend passphrases made of multiple, unrelated words. For example, random combos like “staple-moon-fence-candle” are immune to dictionary attacks if they’re unguessable and not popular memes or in well-known breach lists.
A password like “correcthorse” is (in 2025) as bad as “password123”—it’s too common. But “refinery-stream-drifter-nomad” is good, provided it’s randomly generated.
Password Managers Are Now an Organizational Baseline
The move away from memorizing or writing down complex passphrases means you need password managers in your toolkit. As I pointed out in my post on password managers and data integrity, modern password management tools:
Eliminate reuse by generating random, unique, breach-checked passwords for every system.
Increase the length and randomness of credentials far beyond what humans will remember.
Support compliance and auditing requirements—if you standardize (don’t let employees use their own random apps).
Can even integrate with MFA (multi-factor authentication) for defense in depth.
Critically, as I discuss in the blog post, password manager selection, configuration, and validation are now GxP and audit-relevant. You must document what solutions are allowed (no “bring your own app”), how you test them, and periodic vulnerability and update checks.
What Are the Best Practices for Passwords in 2025?
Let’s lay it out:
Block all dictionary words, product names, and user IDs. Your system must reject any password containing recognizable words, no matter the embellishment.
Screen against breach data and block common patterns. Before accepting a password, check it against up-to-date lists of compromised and weak passwords.
Prioritize password length (minimum 12–16 characters). Random passphrases win. Four or more truly random words (not famous phrases) are vastly superior to gibberish or short “complex” passwords.
Push for password managers. Make one or two IT-validated tools mandatory, make it simple, and do the qualification work. See my advice on password manager selection and qualification.
No forced periodic resets without cause. NIST and ISO 27001 guidance agrees: only reset on suspicion or evidence of compromise, not on a schedule. Forced changes encourage bad habits.
Integrate MFA everywhere possible. Passwords alone are never enough. Multi-factor authentication is the “fail-safe” for inevitable compromise.
Ongoing user education is vital. Explain the risks of dictionary passwords and demonstrate how attack tools work. Show users—and your quality team—why policy isn’t just red tape.
Rewrite Your Password Policy—And Modernize Your Tools
Password security has never been just about meeting a checkbox. In regulated industries, your password policy is a direct reflection of your data integrity posture and audit readiness. Embrace random, unique passphrases. Ban all dictionary words and known patterns. Screen every password against breach data—automatically. Standardize on organization-approved password managers and integrate with MFA whenever possible.
Regulatory expectations from NIST to new draft Annex 11 have joined cybersecurity experts in drawing a clear line: dictionary-word passwords are no longer just bad practice—they’re a compliance landmine.
The draft EU Annex 11 Section 11 “Identity and Access Management” reads like a complete demolition of every lazy access-control practice organizations might have been coasting on for years. Gone are the vague handwaves about “appropriate controls.” The new IAM requirements are explicitly designed to eliminate the shared-account shortcuts and password recycling schemes that have made pharma IT security a running joke among auditors.
The regulatory bar for access management has been raised so high that most existing computerized systems will need major overhauls to comply. Organizations that think a username-password combo and quarterly access reviews will satisfy the new requirements are about to learn some expensive lessons about modern data integrity enforcement.
What Makes This Different from Every Other Access Control Update
The draft Annex 11’s Identity and Access Management section is a complete philosophical shift from “trust but verify” to “prove everything, always.” Where the 2011 version offered generic statements about restricting access to “authorised persons,” the 2025 draft delivers 11 detailed subsections that read like a cybersecurity playbook written by paranoid auditors who’ve spent too much time investigating data integrity failures.
This isn’t incremental improvement. Section 11 transforms IAM from a compliance checkbox into a fundamental pillar of data integrity that touches every aspect of how users interact with GMP systems. The draft makes it explicitly clear that poor access controls are considered violations of data integrity—not just security oversights.
European regulators have decided that the EU needs robust—and arguably more prescriptive—guidance for managing user access in an era where cloud services, remote work, and cyber threats have fundamentally changed the risk landscape. The result is regulatory text that assumes bad actors, compromised credentials, and insider threats as baseline conditions rather than edge cases.
The Eleven Subsections That Will Break Your Current Processes
11.1: Unique Accounts – The Death of Shared Logins
The draft opens with a declaration that will send shivers through organizations still using shared service accounts: “All users should have unique and personal accounts. The use of shared accounts except for those limited to read-only access (no data or settings can be changed), constitute a violation of data integrity”.
This isn’t a suggestion—it’s a flat prohibition with explicit regulatory consequences. Every shared “QC_User” account, every “Production_Shift” login, every “Maintenance_Team” credential becomes a data integrity violation the moment this guidance takes effect. The only exception is read-only accounts that cannot modify data or settings, which means most shared accounts used for batch record reviews, approval workflows, and system maintenance will need complete redesign.
The impact extends beyond just creating more user accounts. This sets out the need to address all the legacy systems that have coasted along for years. There are a lot of filter integrity testers, pH meters and balances, among other systems, that will require some deep views.
Where the 2011 Annex 11 simply required that access changes “should be recorded,” the draft demands “continuous management” with timely granting, modification, and revocation as users “join, change, and end their involvement in GMP activities”. The word “timely” appears to be doing significant regulatory work here—expect inspectors to scrutinize how quickly access is updated when employees change roles or leave the organization.
This requirement acknowledges the reality that modern pharmaceutical operations involve constant personnel changes, contractor rotations, and cross-functional project teams. Static annual access reviews become insufficient when users need different permissions for different projects, temporary elevated access for system maintenance, and immediate revocation when employment status changes. The continuous management standard implies real-time or near-real-time access administration that most organizations currently lack.
The operational implications are clear. It is no longer optional not to integrate HR systems with IT provisioning tools and tie it into your GxP systems. Contractor management processes will require pre-defined access templates and automatic expiration dates. Organizations that treat access management as a periodic administrative task rather than a dynamic business process will find themselves fundamentally out of compliance.
11.3: Certain Identification – The End of Token-Only Authentication
Perhaps the most technically disruptive requirement, Section 11.3 mandates authentication methods that “identify users with a high degree of certainty” while explicitly prohibiting “authentication only by means of a token or a smart card…if this could be used by another user”. This effectively eliminates proximity cards, USB tokens, and other “something you have” authentication methods as standalone solutions.
The regulation acknowledges biometric authentication as acceptable but requires username and password as the baseline, with other methods providing “at least the same level of security”. For organizations that have invested heavily in smart card infrastructure or hardware tokens, this represents a significant technology shift toward multi-factor authentication combining knowledge and possession factors.
The “high degree of certainty” language introduces a subjective standard that will likely be interpreted differently across regulatory jurisdictions. Organizations should expect inspectors to challenge any authentication method that could reasonably be shared, borrowed, or transferred between users. This standard effectively rules out any authentication approach that doesn’t require active user participation—no more swiping someone else’s badge to help them log in during busy periods.
Biometric systems become attractive under this standard, but the draft doesn’t provide guidance on acceptable biometric modalities, error rates, or privacy considerations. Organizations implementing fingerprint, facial recognition, or voice authentication systems will need to document the security characteristics that meet the “high degree of certainty” requirement while navigating European privacy regulations that may restrict biometric data collection.
11.4: Confidential Passwords – Personal Responsibility Meets System Enforcement
The draft’s password confidentiality requirements combine personal responsibility with system enforcement in ways that current pharmaceutical IT environments rarely support. Section 11.4 requires passwords to be “kept confidential and protected from all other users, both at system and at a personal level” while mandating that “passwords received from e.g. a manager, or a system administrator should be changed at the first login, preferably required by the system”1.
This requirement targets the common practice of IT administrators assigning temporary passwords that users may or may not change, creating audit trail ambiguity about who actually performed specific actions. The “preferably required by the system” language suggests that technical controls should enforce password changes rather than relying on user compliance with written procedures.
The personal responsibility aspect extends beyond individual users to organizational accountability. Companies must demonstrate that their password policies, training programs, and technical controls work together to prevent password sharing, writing passwords down, or other practices that compromise authentication integrity. This creates a documentation burden for organizations to prove that their password management practices support data integrity objectives.
11.5: Secure Passwords – Risk-Based Complexity That Actually Works
Rather than mandating specific password requirements, Section 11.5 takes a risk-based approach that requires password rules to be “commensurate with risks and consequences of unauthorised changes in systems and data”. For critical systems, the draft specifies passwords should be “of sufficient length to effectively prevent unauthorised access and contain a combination of uppercase, lowercase, numbers and symbols”.
The regulation prohibits common password anti-patterns: “A password should not contain e.g. words that can be found in a dictionary, the name of a person, a user id, product or organisation, and should be significantly different from a previous password”. This requirement goes beyond basic complexity rules to address predictable password patterns that reduce security effectiveness.
The risk-based approach means organizations must document their password requirements based on system criticality assessments. Manufacturing control systems, quality management databases, and regulatory submission platforms may require different password standards than training systems or general productivity applications. This creates a classification burden where organizations must justify their password requirements through formal risk assessments.
“Sufficient length” and “significantly different” introduce subjective standards that organizations must define and defend. Expect regulatory discussions about whether 8-character passwords meet the “sufficient length” requirement for critical systems, and whether changing a single character constitutes “significantly different” from previous passwords.
11.6: Strong Authentication – MFA for Remote Access
Section 11.6 represents the draft’s most explicit cybersecurity requirement: “Remote authentication on critical systems from outside controlled perimeters, should include multifactor authentication (MFA)”. This requirement acknowledges the reality of remote work, cloud services, and mobile access to pharmaceutical systems while establishing clear security expectations.
The “controlled perimeters” language requires organizations to define their network security boundaries and distinguish between internal and external access. Users connecting from corporate offices, manufacturing facilities, and other company-controlled locations may use different authentication methods than those connecting from home, hotels, or public networks.
“Critical systems” must be defined through risk assessment processes, creating another classification requirement. Organizations must identify which systems require MFA for remote access and document the criteria used for this determination. Laboratory instruments, manufacturing equipment, and quality management systems will likely qualify as critical, but organizations must make these determinations explicitly.
The MFA requirement doesn’t specify acceptable second factors, leaving organizations to choose between SMS codes, authenticator applications, hardware tokens, biometric verification, or other technologies. However, the emphasis on security effectiveness suggests that easily compromised methods like SMS may not satisfy regulatory expectations for critical system access.
11.7: Auto Locking – Administrative Controls for Security Failures
Account lockout requirements in Section 11.7 combine automated security controls with administrative oversight in ways that current pharmaceutical systems rarely implement effectively. The draft requires accounts to be “automatically locked after a pre-defined number of successive failed authentication attempts” with “accounts should only be unlocked by the system administrator after it has been confirmed that this was not part of an unauthorised login attempt or after the risk for such attempt has been removed”.
This requirement transforms routine password lockouts from simple user inconvenience into formal security incident investigations. System administrators cannot simply unlock accounts upon user request—they must investigate the failed login attempts and document their findings before restoring access. For organizations with hundreds or thousands of users, this represents a significant administrative burden that requires defined procedures and potentially additional staffing.
The “pre-defined number” must be established through risk assessment and documented in system configuration. Three failed attempts may be appropriate for highly sensitive systems, while five or more attempts might be acceptable for lower-risk applications. Organizations must justify their lockout thresholds based on balancing security protection with operational efficiency.
“Unauthorised login attempt” investigations require forensic capabilities that many pharmaceutical IT organizations currently lack. System administrators must be able to analyze login patterns, identify potential attack signatures, and distinguish between user errors and malicious activity. This capability implies enhanced logging, monitoring tools, and security expertise that extends beyond traditional IT support functions.
11.8: Inactivity Logout – Session Management That Users Cannot Override
Session management requirements in Section 11.8 establish mandatory timeout controls that users cannot circumvent: “Systems should include an automatic inactivity logout, which logs out a user after a defined period of inactivity. The user should not be able to change the inactivity logout time (outside defined and acceptable limits) or deactivate the functionality”.
The requirement for re-authentication after inactivity logout means users cannot simply resume their sessions—they must provide credentials again, creating multiple authentication points throughout extended work sessions. This approach prevents unauthorized access to unattended workstations while ensuring that long-running analytical procedures or batch processing operations don’t compromise security.
“Defined and acceptable limits” requires organizations to establish timeout parameters based on risk assessment while potentially allowing users some flexibility within security boundaries. A five-minute timeout might be appropriate for systems that directly impact product quality, while 30-minute timeouts could be acceptable for documentation or training applications.
The prohibition on user modification of timeout settings eliminates common workarounds where users extend session timeouts to avoid frequent re-authentication. System configurations must enforce these settings at a level that prevents user modification, requiring administrative control over session management parameters.
Section 11.9 establishes detailed logging requirements that extend far beyond basic audit trail functionality: “Systems should include an access log (separate, or as part of the audit trail) which, for each login, automatically logs the username, user role (if possible, to choose between several roles), the date and time for login, the date and time for logout (incl. inactivity logout)”.
The “separate, or as part of the audit trail” language recognizes that authentication events may need distinct handling from data modification events. Organizations must decide whether to integrate access logs with existing audit trail systems or maintain separate authentication logging capabilities. This decision affects log analysis, retention policies, and regulatory presentation during inspections.
Role logging requirements are particularly significant for organizations using role-based access control systems. Users who can assume different roles during a session (QC analyst, batch reviewer, system administrator) must have their role selections logged with each login event. This requirement supports accountability by ensuring auditors can determine which permissions were active during specific time periods.
The requirement for logs to be “sortable and searchable, or alternatively…exported to a tool which provides this functionality” establishes performance standards for authentication logging systems. Organizations cannot simply capture access events—they must provide analytical capabilities that support investigation, trend analysis, and regulatory review.
11.10: Guiding Principles – Segregation of Duties and Least Privilege
Section 11.10 codifies two fundamental security principles that transform access management from user convenience to risk mitigation: “Segregation of duties, i.e. that users who are involved in GMP activities do not have administrative privileges” and “Least privilege principle, i.e. that users do not have higher access privileges than what is necessary for their job function”.
Segregation of duties eliminates the common practice of granting administrative rights to power users, subject matter experts, or senior personnel who also perform GMP activities. Quality managers cannot also serve as system administrators. Production supervisors cannot have database administrative privileges. Laboratory directors cannot configure their own LIMS access permissions. This separation requires organizations to maintain distinct IT support functions independent from GMP operations.
The least privilege principle requires ongoing access optimization rather than one-time role assignments. Users should receive minimum necessary permissions for their specific job functions, with temporary elevation only when required for specific tasks. This approach conflicts with traditional pharmaceutical access management where users often accumulate permissions over time or receive broad access to minimize support requests.
Implementation of these principles requires formal role definition, access classification, and privilege escalation procedures. Organizations must document job functions, identify minimum necessary permissions, and establish processes for temporary access elevation when users need additional capabilities for specific projects or maintenance activities.
The final requirement establishes ongoing access governance through “recurrent reviews where managers confirm the continued access of their employees in order to detect accesses which should have been changed or revoked during daily operation, but were accidentally forgotten”. This requirement goes beyond periodic access reviews to establish manager accountability for their team’s system permissions.
Manager confirmation creates personal responsibility for access accuracy rather than delegating reviews to IT or security teams. Functional managers must understand what systems their employees access, why those permissions are necessary, and whether access levels remain appropriate for current job responsibilities. This approach requires manager training on system capabilities and access implications.
Role-based access reviews extend the requirement to organizational roles rather than just individual users: “If user accounts are managed by means of roles, these should be subject to the same kind of reviews, where the accesses of roles are confirmed”. Organizations using role-based access control must review role definitions, permission assignments, and user-to-role mappings with the same rigor applied to individual account reviews.
Documentation and action requirements ensure that reviews produce evidence and corrections: “The reviews should be documented, and appropriate action taken”. Organizations cannot simply perform reviews—they must record findings, document decisions, and implement access modifications identified during the review process.
Risk-based frequency allows organizations to adjust review cycles based on system criticality: “The frequency of these reviews should be commensurate with the risks and consequences of changes in systems and data made by unauthorised individuals”. Critical manufacturing systems may require monthly reviews, while training systems might be reviewed annually.
How This Compares to 21 CFR Part 11 and Current Best Practices
The draft Annex 11’s Identity and Access Management requirements represent a significant advancement over 21 CFR Part 11, which addressed access control through basic authority checks and user authentication rather than comprehensive identity management. Part 11’s requirement for “at least two distinct identification components” becomes the foundation for much more sophisticated authentication and access control frameworks.
Multi-factor authentication requirements in the draft Annex 11 exceed Part 11 expectations by mandating MFA for remote access to critical systems, while Part 11 remains silent on multi-factor approaches. This difference reflects 25 years of cybersecurity evolution and acknowledges that username-password combinations provide insufficient protection for modern threat environments.
Current data integrity best practices have evolved toward comprehensive access management, risk-based authentication, and continuous monitoring—approaches that the draft Annex 11 now mandates rather than recommends. Organizations following ALCOA+ principles and implementing robust access controls will find the new requirements align with existing practices, while those relying on minimal compliance approaches will face significant gaps.
The Operational Reality of Implementation
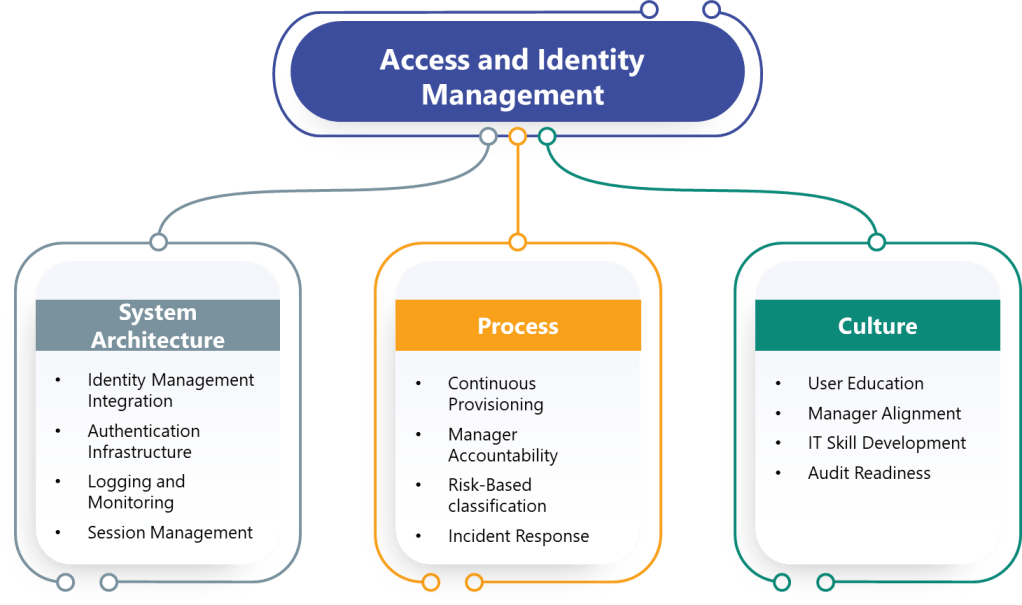
System Architecture Changes
Most pharmaceutical computerized systems were designed assuming manual access management and periodic reviews would satisfy regulatory requirements. The draft Annex 11 requirements will force fundamental architecture changes including:
Identity Management Integration: Manufacturing execution systems, laboratory information management systems, and quality management platforms must integrate with centralized identity management systems to support continuous access management and role-based controls.
Authentication Infrastructure: Organizations must deploy multi-factor authentication systems capable of supporting diverse user populations, remote access scenarios, and integration with existing applications.
Logging and Monitoring: Enhanced access logging requirements demand centralized log management, analytical capabilities, and integration between authentication systems and audit trail infrastructure.
Session Management: Applications must implement configurable session timeout controls, prevent user modification of security settings, and support re-authentication without disrupting long-running processes.
Process Reengineering Requirements
The regulatory requirements will force organizations to redesign fundamental access management processes:
Continuous Provisioning: HR onboarding, role changes, and termination processes must trigger immediate access modifications rather than waiting for periodic reviews.
Manager Accountability: Access review processes must shift from IT-driven activities to manager-driven confirmations with documented decision-making and corrective actions.
Risk-Based Classification: Organizations must classify systems based on criticality, define access requirements accordingly, and maintain documentation supporting these determinations.
Incident Response: Account lockout events must trigger formal security investigations rather than simple password resets, requiring enhanced forensic capabilities and documented procedures.
Manager Training: Functional managers must understand system capabilities, access implications, and review responsibilities rather than delegating access decisions to IT teams.
User Education: Password security, MFA usage, and session management practices require user training programs that emphasize data integrity implications rather than just security compliance.
IT Skill Development: System administrators must develop security investigation capabilities, risk assessment skills, and regulatory compliance expertise beyond traditional technical support functions.
Audit Readiness: Organizations must prepare to demonstrate access control effectiveness through documentation, metrics, and investigative capabilities during regulatory inspections.
Strategic Implementation Approach
The Annex 11 Draft is just taking good cybersecurity and enshrining it more firmly in the GMPs. Organizations should not wait for the effective version to implement. Get that budget prioritized and start now.
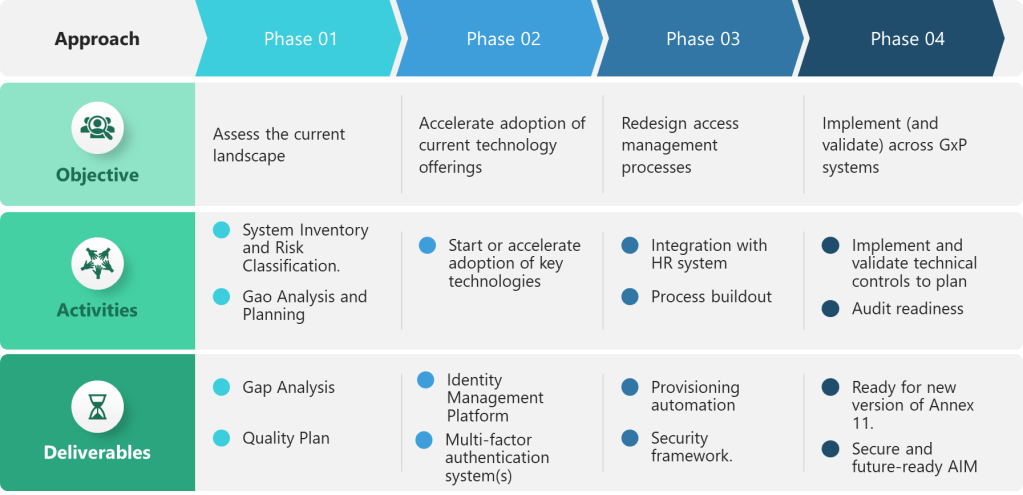
Phase 1: Assessment and Classification
Organizations should begin with comprehensive assessment of current access control practices against the new requirements:
System Inventory: Catalog all computerized systems used in GMP activities, identifying shared accounts, authentication methods, and access control capabilities.
Risk Classification: Determine which systems qualify as “critical” requiring enhanced authentication and access controls.
Gap Analysis: Compare current practices against each subsection requirement, identifying technical, procedural, and training gaps.
Compliance Timeline: Develop implementation roadmap aligned with expected regulatory effective dates and system upgrade cycles.
Phase 2: Infrastructure Development
Focus on foundational technology changes required to support the new requirements:
Identity Management Platform: Deploy or enhance centralized identity management systems capable of supporting continuous provisioning and role-based access control.
Multi-Factor Authentication: Implement MFA systems supporting diverse authentication methods and integration with existing applications.
Enhanced Logging: Deploy log management platforms capable of aggregating, analyzing, and presenting access events from distributed systems.
Session Management: Upgrade applications to support configurable timeout controls and prevent user modification of security settings.
Phase 3: Process Implementation
Redesign access management processes to support continuous management and enhanced accountability:
Provisioning Automation: Integrate HR systems with IT provisioning tools to support automatic access changes based on employment events.
Manager Accountability: Train functional managers on access review responsibilities and implement documented review processes.
Security Incident Response: Develop procedures for investigating account lockouts and documenting security findings.
Audit Trail Integration: Ensure access events are properly integrated with existing audit trail review and batch release processes.
Phase 4: Validation and Documentation
When the Draft becomes effective you’ll be ready to complete validation activities demonstrating compliance with the new requirements:
Access Control Testing: Validate that technical controls prevent unauthorized access, enforce authentication requirements, and log security events appropriately.
Process Verification: Demonstrate that access management processes support continuous management, manager accountability, and risk-based reviews.
Training Verification: Document that personnel understand their responsibilities for password security, session management, and access control compliance.
Audit Readiness: Prepare documentation, metrics, and investigative capabilities required to demonstrate compliance during regulatory inspections.
The Competitive Advantage of Early Implementation
Organizations that proactively implement the draft Annex 11 IAM requirements will gain significant advantages beyond regulatory compliance:
Enhanced Security Posture: The access control improvements provide protection against cyber threats, insider risks, and accidental data compromise that extend beyond GMP applications to general IT security.
Operational Efficiency: Automated provisioning, role-based access, and centralized identity management reduce administrative overhead while improving access accuracy.
Audit Confidence: Comprehensive access logging, manager accountability, and continuous management provide evidence of control effectiveness that regulators and auditors will recognize.
Digital Transformation Enablement: Modern identity and access management infrastructure supports cloud adoption, mobile access, and advanced analytics initiatives that drive business value.
Risk Mitigation: Enhanced access controls reduce the likelihood of data integrity violations, security incidents, and regulatory findings that can disrupt operations and damage reputation.
Looking Forward: The End of Security Theater
The draft Annex 11’s Identity and Access Management requirements represent the end of security theater in pharmaceutical access control. Organizations can no longer satisfy regulatory expectations through generic policies and a reliance on periodic reviews to provide the appearance of control without actual security effectiveness.
The new requirements assume that user access is a continuous risk requiring active management, real-time monitoring, and ongoing verification. This approach aligns with modern cybersecurity practices while establishing regulatory expectations that reflect the actual threat environment facing pharmaceutical operations.
Implementation success will require significant investment in technology infrastructure, process reengineering, and organizational change management. However, organizations that embrace these requirements as opportunities for security improvement rather than compliance burdens will build competitive advantages that extend far beyond regulatory satisfaction.
The transition period between now and the expected 2026 effective date provides a ideal window for organizations to assess their current practices, develop implementation strategies, and begin the technical and procedural changes required for compliance. Organizations that delay implementation risk finding themselves scrambling to achieve compliance while their competitors demonstrate regulatory leadership through proactive adoption.
For pharmaceutical organizations serious about data integrity, operational security, and regulatory compliance, the draft Annex 11 IAM requirements aren’t obstacles to overcome—they’re the roadmap to building access control practices worthy of the products and patients we serve. The only question is whether your organization will lead this transformation or follow in the wake of those who do.
Requirement
Current Annex 11 (2011)
Draft Annex 11 Section 11 (2025)
21 CFR Part 11
User Account Management
Basic – creation, change, cancellation should be recorded
Continuous management – grant, modify, revoke as users join/change/leave
Basic user management, creation/change/cancellation recorded
Authentication Methods
Physical/logical controls, pass cards, personal codes with passwords, biometrics
Username + password or equivalent (biometrics); tokens/smart cards alone insufficient
At least two distinct identification components (ID code + password)
Password Requirements
Not specified in detail
Secure passwords enforced by systems, length/complexity based on risk, dictionary words prohibited
Ready or not, the EU’s draft revision of Annex 11 is moving toward finalization, and its brand-new Section 13 on electronic signatures is a wake-up call for anyone still treating digital authentication as just Part 11 with an accent. In this post I will take a deep dive into what’s changing, why it matters, and how to keep your quality system out of the regulatory splash zone.
Section 13 turns electronic signatures from a check-the-box formality into a risk-based, security-anchored discipline. Think multi-factor authentication, time-zone stamps, hybrid wet-ink safeguards, and explicit “non-repudiation” language—all enforced at the same rigor as system login. If your current SOPs still assume username + password = done, it’s time to start planning some improvements.
Why the Rewrite?
Tech has moved on: Biometric ID, cloud PaaS, and federated identity management were sci-fi when the 2011 Annex 11 dropped.
Threat landscape: Ransomware and credential stuffing didn’t exist at today’s scale. Regulators finally noticed.
Global convergence: The FDA’s Computer Software Assurance (CSA) draft and PIC/S data-integrity guides pushed the EU to level up.
Must equal or exceed login strength; first sign = full re-auth, subsequent signs = pwd/biometric; smart-card-only = banned
Two identification components
Forces MFA or biometrics; goodbye “remember me” shortcuts
Time & Time-Zone
Date + time (manual OK)
Auto-captured and time-zone logged
Date + time (no TZ)
Multisite ops finally get defensible chronology
Signature Meaning Prompt
Not required
System must ask user for purpose (approve, review…)
Required but less prescriptive
Eliminates “mystery clicks” that auditors love to exploit
Manifestation Elements
Minimal
Full name, username, role, meaning, date/time/TZ
Name, date, meaning
Closes attribution gaps; boosts ALCOA+ “Legible”
Indisputability Clause
“Same impact”
Explicit non-repudiation mandate
Equivalent legal weight
Sets the stage for eIDAS/federated ID harmonization
Record Linking After Change
Permanent link
If record altered post-sign, signature becomes void/flagged
Link cannot be excised
Ends stealth edits after approval
Hybrid Wet-Ink Control
Silent
Hash code or similar to break link if record changes
Silent
Lets you keep occasional paper without tanking data integrity
Open Systems / Trusted Services
Silent
Must comply with “national/international trusted services” (read: eIDAS)
Extra controls, but legacy wording
Validates cloud signing platforms out of the box
The Implications
Multi-Factor Authentication (MFA) Is Now Table Stakes
Because the draft explicitly bars any authentication method that relies solely on a smart card or a static PIN, every electronic signature now has to be confirmed with an additional, independent factor—such as a password, biometric scan, or time-limited one-time code—so that the credential used to apply the signature is demonstrably different from the one that granted the user access to the system in the first place.
Time-Zone Logging Kills Spreadsheet Workarounds
One of the more subtle but critical updates in Draft Annex 11’s Section 13.4 is the explicit requirement for automatic logging of the time zone when electronic signatures are applied. Unlike previous guidance—whether under the 2011 Annex 11 or 21 CFR Part 11—that only mandated the capture of date and time (often allowing manual entry or local system time), the draft stipulates that systems must automatically capture the precise time and associated time zone for each signature event. This seemingly small detail has monumental implications for data integrity, traceability, and regulatory compliance. Why does this matter? For global pharmaceutical operations spanning multiple time zones, manual or local-only timestamps often create ambiguous or conflicting audit trails, leading to discrepancies in event sequencing. Companies relying on spreadsheets or legacy systems that do not incorporate time zone information effectively invite errors where a signature in one location appears to precede an earlier event simply due to zone differences. This ambiguity can undermine the “Contemporaneous” and “Enduring” principles of ALCOA+, principles the draft Annex 11 explicitly reinforces throughout electronic signature requirements. By mandating automated, time zone-aware timestamping, Draft Annex 11 Section 13.4 ensures that electronic signature records maintain a defensible and standardized chronology across geographies, eliminating the need for cumbersome manual reconciliation or retrospective spreadsheet corrections. This move not only tightens compliance but also supports modern, centralized data review and analytics where uniform timestamping is essential. If your current systems or SOPs rely on manual date/time entry or overlook time zone logging, prepare for significant system and procedural updates to meet this enhanced expectation once the draft Annex 11 is finalized. .
Hybrid Records Are Finally Codified
If you still print a batch record for wet-ink QA approval, Section 13.9 lets you keep the ritual—but only if a cryptographic hash or similar breaks when someone tweaks the underlying PDF. Expect a flurry of DocuSign-scanner-hash utilities.
Open-System Signatures Shift Liability
Draft Annex 11’s Section 13.2 represents perhaps the most strategically significant change in electronic signature liability allocation since 21 CFR Part 11 was published in 1997. The provision states that “Where the system owner does not have full control of system accesses (open systems), or where required by other legislation, electronic signatures should, in addition, meet applicable national and international requirements, such as trusted services”. This seemingly simple sentence fundamentally reshapes liability relationships in modern pharmaceutical IT architectures.
Defining the Open System Boundary
The draft Annex 11 adopts the 21 CFR Part 11 definition of open systems—environments where system owners lack complete control over access and extends it into contemporary cloud, SaaS, and federated identity scenarios. Unlike the original Part 11 approach, which merely required “additional measures such as document encryption and use of appropriate digital signature standards”, Section 13.2 creates a positive compliance obligation by mandating adherence to “trusted services” frameworks.
This distinction is critical: while Part 11 treats open systems as inherently risky environments requiring additional controls, draft Annex 11 legitimizes open systems provided they integrate with qualified trust service providers. Organizations no longer need to avoid cloud-based signature services; instead, they must ensure those services meet eIDAS-qualified standards or equivalent national frameworks.
The Trusted Services Liability Transfer
Section 13.2’s reference to “trusted services” directly incorporates European eIDAS Regulation 910/2014 into pharmaceutical GMP compliance, creating what amounts to a liability transfer mechanism. Under eIDAS, Qualified Trust Service Providers (QTSPs) undergo rigorous third-party audits, maintain certified infrastructure, and provide legal guarantees about signature validity and non-repudiation. When pharmaceutical companies use eIDAS-qualified signature services, they effectively transfer signature validity liability from their internal systems to certified external providers.
This represents a fundamental shift from the 21 CFR Part 11 closed-system preference, where organizations maintained complete control over signature infrastructure but also bore complete liability for signature failures. Draft Annex 11 acknowledges that modern pharmaceutical operations often depend on cloud service providers, federated authentication systems, and external trust services—and provides a regulatory pathway to leverage these technologies while managing liability exposure.
Practical Implications for SaaS Platforms
The most immediate impact affects organizations using Software-as-a-Service platforms for clinical data management, quality management, or document management. Under current Annex 11 and Part 11, these systems often require complex validation exercises to demonstrate signature integrity, with pharmaceutical companies bearing full responsibility for signature validity even when using external platforms.
Section 13.2 changes this dynamic by validating reliance on qualified trust services. Organizations using platforms like DocuSign, Adobe Sign, or specialized pharmaceutical SaaS providers can now satisfy Annex 11 requirements by ensuring their chosen platforms integrate with eIDAS-qualified signature services. The pharmaceutical company’s validation responsibility shifts from proving signature technology integrity to verifying trust service provider qualifications and proper integration.
Integration with Identity and Access Management
Draft Annex 11’s Section 11 (Identity and Access Management) works in conjunction with Section 13.2 to support federated identity scenarios common in modern pharmaceutical operations. Organizations can now implement single sign-on (SSO) systems with external identity providers, provided the signature components integrate with trusted services. This enables scenarios where employees authenticate through corporate Active Directory systems but execute legally binding signatures through eIDAS-qualified providers.
The liability implications are significant: authentication failures become the responsibility of the identity provider (within contractual limits), while signature validity becomes the responsibility of the qualified trust service provider. The pharmaceutical company retains responsibility for proper system integration and user access controls, but shares technical implementation liability with certified external providers.
Cloud Service Provider Risk Allocation
For organizations using cloud-based LIMS, MES, or quality management systems, Section 13.2 provides regulatory authorization to implement signature services hosted entirely by external providers. Cloud service providers offering eIDAS-compliant signature services can contractually accept liability for signature technical implementation, cryptographic integrity, and legal validity—provided they maintain proper trust service qualifications.
This risk allocation addresses a long-standing concern in pharmaceutical cloud adoption: the challenge of validating signature infrastructure owned and operated by external parties. Under Section 13.2, organizations can rely on qualified trust service provider certifications rather than conducting detailed technical validation of cloud provider signature implementations.
Harmonization with Global Standards
Section 13.2’s “national and international requirements” language extends beyond eIDAS to encompass other qualified electronic signature frameworks. This includes Swiss ZertES standards and Canadian digital signature regulations,. Organizations operating globally can implement unified signature platforms that satisfy multiple regulatory requirements through single trusted service provider integrations.
The practical effect is regulatory arbitrage: organizations can choose signature service providers based on the most favorable combination of technical capabilities, cost, and regulatory coverage, rather than being constrained by local regulatory limitations.
Supplier Assessment Transformation
Draft Annex 11’s Section 7 (Supplier and Service Management) requires comprehensive supplier assessment for computerized systems. However, Section 13.2 creates a qualified exception for eIDAS-certified trust service providers: organizations can rely on third-party certification rather than conducting independent technical assessments of signature infrastructure.
This significantly reduces supplier assessment burden for signature services. Instead of auditing cryptographic implementations, hardware security modules, and signature validation algorithms, organizations can verify trust service provider certifications and assess integration quality. The result: faster implementation cycles and reduced validation costs for signature-enabled systems.
Audit Trail Integration Considerations
The liability shift enabled by Section 13.2 affects audit trail management requirements detailed in draft Annex 11’s expanded Section 12 (Audit Trails). When signature events are managed by external trust service providers, organizations must ensure signature-related audit events are properly integrated with internal audit trail systems while maintaining clear accountability boundaries.
Qualified trust service providers typically provide comprehensive signature audit logs, but organizations remain responsible for correlation with business process audit trails. This creates shared audit trail management where signature technical events are managed externally but business context remains internal responsibility.
Competitive Advantages of Early Adoption
Organizations that proactively implement Section 13.2 requirements gain several strategic advantages:
Reduced Infrastructure Costs: Elimination of internal signature infrastructure maintenance and validation overhead
Enhanced Security: Leverage specialized trust service provider security expertise and certified infrastructure
Global Scalability: Unified signature platforms supporting multiple regulatory jurisdictions through single provider relationships
Accelerated Digital Transformation: Faster deployment of signature-enabled processes through validated external services
Risk Transfer: Contractual liability allocation with qualified external providers rather than complete internal risk retention
Section 13.2 transforms open system electronic signatures from compliance challenges into strategic enablers of digital pharmaceutical operations. By legitimizing reliance on qualified trust services, the draft Annex 11 enables organizations to leverage best-in-class signature technologies while managing regulatory compliance and liability exposure through proven external partnerships. The result: more secure, cost-effective, and globally scalable electronic signature implementations that support advanced digital quality management systems.
How to Get Ahead (Instead of Playing Cleanup)
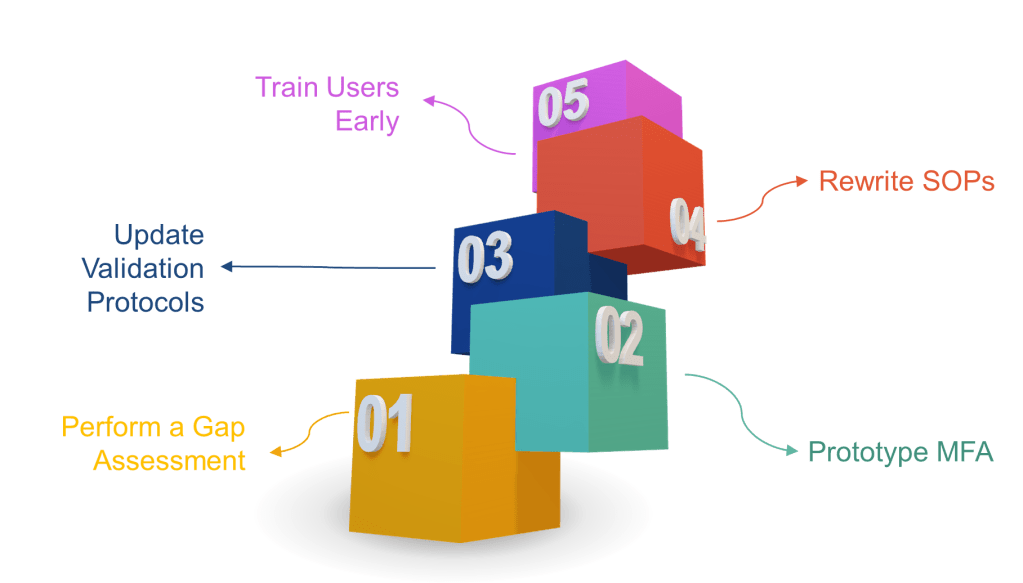
Perform a gap assessment now—map every signature point to the new rules.
Prototype MFA in your eDMS or MES. If users scream about friction, remind them that ransomware is worse.
Update validation protocols to include time-zone, hybrid record, and non-repudiation tests.
Rewrite SOPs to include signature-meaning prompts and periodic access-right recertification.
Train users early. A 30-second “why you must re-authenticate” explainer video beats 300 deviations later.
Final Thoughts
The draft Annex 11 doesn’t just tweak wording—it yanks electronic signatures into the 2020s. Treat Section 13 as both a compliance obligation and an opportunity to slash latent data-integrity risk. Those who adapt now will cruise through 2026/2027 inspections while the laggards scramble for remediation budgets.
I have a few concerns about George Tidmarsh’s recent appointment as director of the FDA’s Center for Drug Evaluation and Research (CDER) which fits into wider concerns about the current administration. As usual in this adminstration, this boils down to a tendency to fringe medicine and vaccine denial adjacent thinking.
Association with “fringe” medical publishing: Tidmarsh contributed to the Journal of the Academy of Public Health, which Bloomberg and other outlets have described as a “fringe medical journal” connected to a conservative nonprofit.
Criticism of COVID-19 public health policy: Tidmarsh has openly criticized the government’s handling of the COVID-19 pandemic. On the “Derate the Hate” podcast, he lamented what he saw as political polarization and lack of “academic freedom” in pandemic-era policy discussions, and suggested openness to the theory of a lab origin for the coronavirus.
I also find a degree of irony in Tidmarsh’s leadership roles in several biotech and pharmaceutical companies—including as founder and CEO of Horizon Therapeutics and Threshold Pharmaceuticals. RFKJr, and others have spent a lot of time criticizing the revolving door between industry and government regulation. And here we have a great example of that revolving door.
In any other administration, Tidmarsh would be concerning due to his connections to fringe science. In this administration he is another worrisome personnel decision given authority.