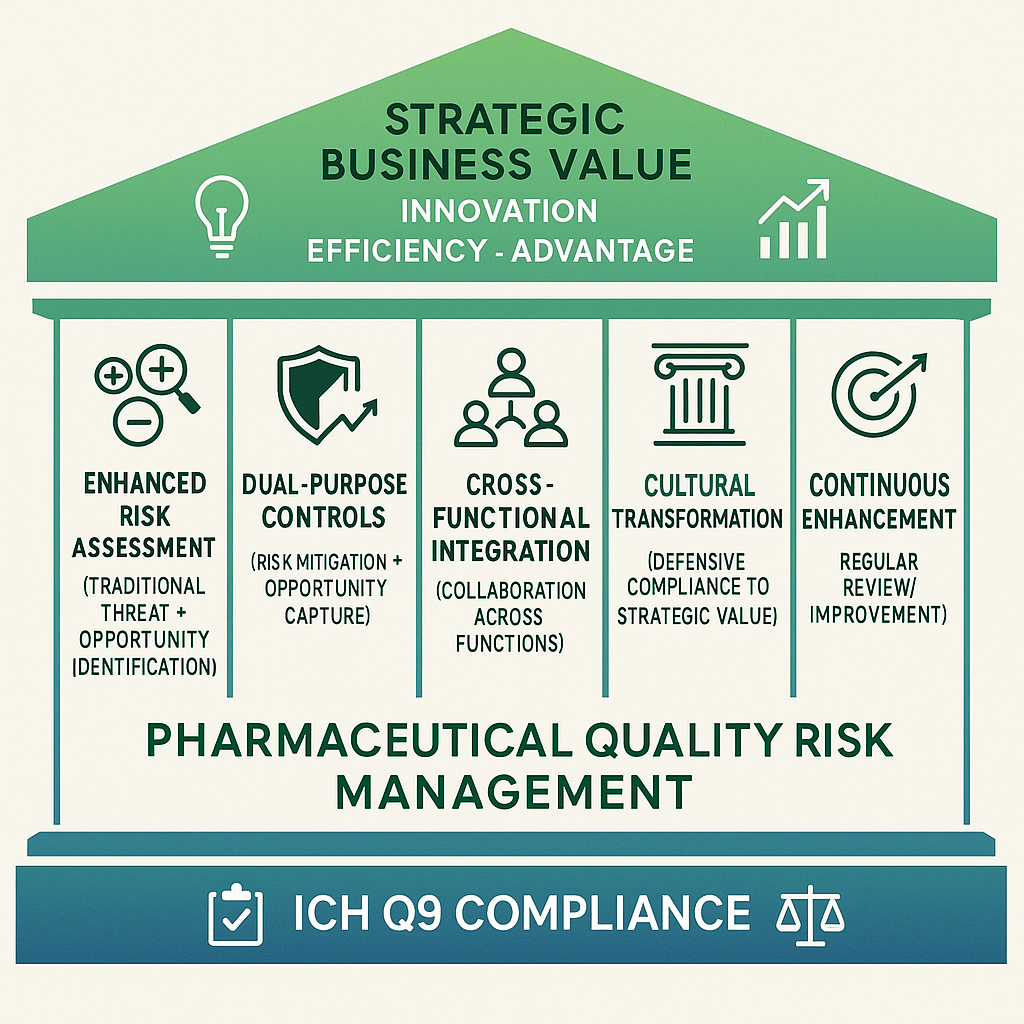
The pharmaceutical industry has long operated under a defensive mindset when it comes to risk management. We identify what could go wrong, assess the likelihood and impact of failure modes, and implement controls to prevent or mitigate negative outcomes. This approach, while necessary and required by ICH Q9, represents only half the risk equation. What our quality risk management program could become not just a compliance necessity, but a strategic driver of innovation, efficiency, and competitive advantage?
Enter the ISO 31000 perspective on risk—one that recognizes risk as “the effect of uncertainty on objectives,” where that effect can be positive, negative, or both. This broader definition opens up transformative possibilities for how we approach quality risk management in pharmaceutical manufacturing. Rather than solely focusing on preventing bad things from happening, we can start identifying and capitalizing on good things that might occur.
The Evolution of Risk Thinking in Pharmaceuticals
For decades, our industry’s risk management approach has been shaped by regulatory necessity and liability concerns. The introduction of ICH Q9 in 2005—and its recent revision in 2023—provided a structured framework for quality risk management that emphasizes scientific knowledge, proportional formality, and patient protection. This framework has served us well, establishing systematic approaches to risk assessment, control, communication, and review.
However, the updated ICH Q9(R1) recognizes that we’ve been operating with significant blind spots. The revision addresses issues including “high levels of subjectivity in risk assessments,” “failing to adequately manage supply and product availability risks,” and “lack of clarity on risk-based decision-making”. These challenges suggest that our traditional approach to risk management, while compliant, may not be fully leveraging the strategic value that comprehensive risk thinking can provide.
The ISO 31000 standard offers a complementary perspective that can address these gaps. By defining risk as uncertainty’s effect on objectives—with explicit recognition that this effect can create opportunities as well as threats—ISO 31000 provides a framework for risk management that is inherently more strategic and value-creating.
Understanding Risk as Opportunity in the Pharmaceutical Context
Lot us start by establishing a clear understanding of what “positive risk” or “opportunity” means in our context. In pharmaceutical quality management, opportunities are uncertain events or conditions that, if they occur, would enhance our ability to achieve quality objectives beyond our current expectations.
Consider these examples:
Manufacturing Process Opportunities: A new analytical method validates faster than anticipated, allowing for reduced testing cycles and increased throughput. The uncertainty around validation timelines created an opportunity that, when realized, improved operational efficiency while maintaining quality standards.
Supply Chain Opportunities: A raw material supplier implements process improvements that result in higher-purity ingredients at lower cost. This positive deviation from expected quality created opportunities for enhanced product stability and improved margins.
Technology Integration Opportunities: Implementation of process analytical technology (PAT) tools not only meets their intended monitoring purpose but reveals previously unknown process insights that enable further optimization opportunities.
Regulatory Opportunities: A comprehensive quality risk assessment submitted as part of a regulatory filing demonstrates such thorough understanding of the product and process that regulators grant additional manufacturing flexibility, creating opportunities for more efficient operations.
These scenarios illustrate how uncertainty—the foundation of all risk—can work in our favor when we’re prepared to recognize and capitalize on positive outcomes.
The Strategic Value of Opportunity-Based Risk Management
Integrating opportunity recognition into your quality risk management program delivers value across multiple dimensions:
Enhanced Innovation Capability
Traditional risk management often creates conservative cultures where “safe” decisions are preferred over potentially transformative ones. By systematically identifying and evaluating opportunities, we can make more balanced decisions that account for both downside risks and upside potential. This leads to greater willingness to explore innovative approaches to quality challenges while maintaining appropriate risk controls.
Improved Resource Allocation
When we only consider negative risks, we tend to over-invest in protective measures while under-investing in value-creating activities. Opportunity-oriented risk management helps optimize resource allocation by identifying where investments might yield unexpected benefits beyond their primary purpose.
Strengthened Competitive Position
Companies that effectively identify and capitalize on quality-related opportunities can develop competitive advantages through superior operational efficiency, faster time-to-market, enhanced product quality, or innovative approaches to regulatory compliance.
Cultural Transformation
Perhaps most importantly, embracing opportunities transforms the perception of risk management from a necessary burden to a strategic enabler. This cultural shift encourages proactive thinking, innovation, and continuous improvement throughout the organization.
Mapping ISO 31000 Principles to ICH Q9 Requirements
The beauty of integrating ISO 31000’s opportunity perspective with ICH Q9 compliance lies in their fundamental compatibility. Both frameworks emphasize systematic, science-based approaches to risk management with proportional formality based on risk significance. The key difference is scope—ISO 31000’s broader definition of risk naturally encompasses opportunities alongside threats.
Risk Assessment Enhancement
ICH Q9 requires risk assessment to include hazard identification, analysis, and evaluation. The ISO 31000 approach enhances this by expanding identification beyond failure modes to include potential positive outcomes. During hazard analysis and risk assessment (HARA), we can systematically ask not only “what could go wrong?” but also “what could go better than expected?” and “what positive outcomes might emerge from this uncertainty?”
For example, when assessing risks associated with implementing a new manufacturing technology, traditional ICH Q9 assessment would focus on potential failures, integration challenges, and validation risks. The enhanced approach would also identify opportunities for improved process understanding, unexpected efficiency gains, or novel approaches to quality control that might emerge during implementation.
Risk Control Expansion
ICH Q9’s risk control phase traditionally focuses on risk reduction and risk acceptance. The ISO 31000 perspective adds a third dimension: opportunity enhancement. This involves implementing controls or strategies that not only mitigate negative risks but also position the organization to capitalize on positive uncertainties should they occur.
Consider controls designed to manage analytical method transfer risks. Traditional controls might include extensive validation studies, parallel testing, and contingency procedures. Opportunity-enhanced controls might also include structured data collection protocols designed to identify process insights, cross-training programs that build broader organizational capabilities, or partnerships with equipment vendors that could lead to preferential access to new technologies.
Risk Communication and Opportunity Awareness
ICH Q9 emphasizes the importance of risk communication among stakeholders. When we expand this to include opportunity communication, we create organizational awareness of positive possibilities that might otherwise go unrecognized. This enhanced communication helps ensure that teams across the organization are positioned to identify and report positive deviations that could represent valuable opportunities.
Risk Review and Opportunity Capture
The risk review process required by ICH Q9 becomes more dynamic when it includes opportunity assessment. Regular reviews should evaluate not only whether risk controls remain effective, but also whether any positive outcomes have emerged that could be leveraged for further benefit. This creates a feedback loop that continuously enhances both risk management and opportunity realization.
Implementation Framework
Implementing opportunity-based risk management within your existing ICH Q9 program requires systematic integration rather than wholesale replacement. Here’s a practical framework for making this transition:
Phase 1: Assessment and Planning
Begin by evaluating your current risk management processes to identify integration points for opportunity assessment. Review existing risk assessments to identify cases where positive outcomes might have been overlooked. Establish criteria for what constitutes a meaningful opportunity in your context—this might include potential cost savings, quality improvements, efficiency gains, or innovation possibilities above defined thresholds.
Key activities include:
- Mapping current risk management processes against ISO 31000 principles
- Perform a readiness evaluation
- Training risk management teams on opportunity identification techniques
- Developing templates and tools that prompt opportunity consideration
- Establishing metrics for tracking opportunity identification and realization
Readiness Evaluation
Before implementing opportunity-based risk management, conduct a thorough assessment of organizational readiness and capability. This includes evaluating current risk management maturity, cultural factors that might support or hinder adoption, and existing processes that could be enhanced.
Key assessment areas include:
- Current risk management process effectiveness and consistency
- Organizational culture regarding innovation and change
- Leadership support for expanded risk management approaches
- Available resources for training and process enhancement
- Existing cross-functional collaboration capabilities
Phase 2: Process Integration
Systematically integrate opportunity assessment into your existing risk management workflows. This doesn’t require new procedures—rather, it involves enhancing existing processes to ensure opportunity identification receives appropriate attention alongside threat assessment.
Modify risk assessment templates to include opportunity identification sections. Train teams to ask opportunity-focused questions during risk identification sessions. Develop criteria for evaluating opportunity significance using similar approaches to threat assessment—considering likelihood, impact, and detectability.
Update risk control strategies to include opportunity enhancement alongside risk mitigation. This might involve designing controls that serve dual purposes or implementing monitoring systems that can detect positive deviations as well as negative ones.
This is the phase I am currently working through. Make sure to do a pilot program!
Pilot Program Development
Start with pilot programs in areas where opportunities are most likely to be identified and realized. This might include new product development projects, technology implementation initiatives, or process improvement activities where uncertainty naturally creates both risks and opportunities.
Design pilot programs to:
- Test opportunity identification and evaluation methods
- Develop organizational capability and confidence
- Create success stories that support broader adoption
- Refine processes and tools based on practical experience
Phase 3: Cultural Integration
The success of opportunity-based risk management ultimately depends on cultural adoption. Teams need to feel comfortable identifying and discussing positive possibilities without being perceived as overly optimistic or insufficiently rigorous.
Establish communication protocols that encourage opportunity reporting alongside issue escalation. Recognize and celebrate cases where teams successfully identify and capitalize on opportunities. Incorporate opportunity realization into performance metrics and success stories.
Scaling and Integration Strategy
Based on pilot program results, develop a systematic approach for scaling opportunity-based risk management across the organization. This should include timelines, resource requirements, training programs, and change management strategies.
Consider factors such as:
- Process complexity and risk management requirements in different areas
- Organizational change capacity and competing priorities
- Resource availability and investment requirements
- Integration with other improvement and innovation initiatives
Phase 4: Continuous Enhancement
Like all aspects of quality risk management, opportunity integration requires continuous improvement. Regular assessment of the program’s effectiveness in identifying and capitalizing on opportunities helps refine the approach over time.
Conduct periodic reviews of opportunity identification accuracy—are teams successfully recognizing positive outcomes when they occur? Evaluate opportunity realization effectiveness—when opportunities are identified, how successfully does the organization capitalize on them? Use these insights to enhance training, processes, and organizational support for opportunity-based risk management.
Long-term Sustainability Planning
Ensure that opportunity-based risk management becomes embedded in organizational culture and processes rather than remaining dependent on individual champions or special programs. This requires systematic integration into standard operating procedures, performance metrics, and leadership expectations.
Plan for:
- Ongoing training and capability development programs
- Regular assessment and continuous improvement of opportunity identification processes
- Integration with career development and advancement criteria
- Long-term resource allocation and organizational support
Tools and Techniques for Opportunity Integration
Include a Success Mode and Benefits Analysis in your FMEA (Failure Mode and Effects Analysis)
Traditional FMEA focuses on potential failures and their effects. Opportunity-enhanced FMEA includes “Success Mode and Benefits Analysis” (SMBA) that systematically identifies potential positive outcomes and their benefits. For each process step, teams assess not only what could go wrong, but also what could go better than expected and how to position the organization to benefit from such outcomes.
A Success Mode and Benefits Analysis (SMBA) is the positive complement to the traditional Failure Mode and Effects Analysis (FMEA). While FMEA identifies where things can go wrong and how to prevent or mitigate failures, SMBA systematically evaluates how things can go unexpectedly right—helping organizations proactively capture, enhance, and realize benefits that arise from process successes, innovations, or positive deviations.
What Does a Success Mode and Benefits Analysis Look Like?
The SMBA is typically structured as a table or worksheet with a format paralleling the FMEA, but with a focus on positive outcomes and opportunities. A typical SMBA process includes the following columns and considerations:
| Step/Column | Description |
|---|---|
| Process Step/Function | The specific process, activity, or function under investigation. |
| Success Mode | Description of what could go better than expected or intended—what’s the positive deviation? |
| Benefits/Effects | The potential beneficial effects if the success mode occurs (e.g., improved yield, faster cycle, enhanced quality, regulatory flexibility). |
| Likelihood (L) | Estimated probability that the success mode will occur. |
| Magnitude of Benefit (M) | Qualitative or quantitative evaluation of how significant the benefit would be (e.g., minor, moderate, major; or by quantifiable metrics). |
| Detectability | Can the opportunity be spotted early? What are the triggers or signals of this benefit occurring? |
| Actions to Capture/Enhance | Steps or controls that could help ensure the success is recognized and benefits are realized (e.g., monitoring plans, training, adaptation of procedures). |
| Benefit Priority Number (BPN) | An optional calculated field (e.g., L × M) to help the team prioritize follow-up actions. |
- Proactive Opportunity Identification: Instead of waiting for positive results to emerge, the process prompts teams to seek out “what could go better than planned?”.
- Systematic Benefit Analysis: Quantifies or qualifies benefits just as FMEA quantifies risk.
- Follow-Up Actions: Establishes ways to amplify and institutionalize successes.
When and How to Use SMBA
- Use SMBA alongside FMEA during new technology introductions, process changes, or annual reviews.
- Integrate into cross-functional risk assessments to balance risk aversion with innovation.
- Use it to foster a culture that not just “prevents failure,” but actively “captures opportunity” and learns from success.
Opportunity-Integrated Risk Matrices
Traditional risk matrices plot likelihood versus impact for negative outcomes. Enhanced matrices include separate quadrants or scales for positive outcomes, allowing teams to visualize both threats and opportunities in the same framework. This provides a more complete picture of uncertainty and helps prioritize actions based on overall risk-opportunity balance.
Scenario Planning with Upside Cases
While scenario planning typically focuses on “what if” situations involving problems, opportunity-oriented scenario planning includes “what if” situations involving unexpected successes. This helps teams prepare to recognize and capitalize on positive outcomes that might otherwise be missed.
Innovation-Focused Risk Assessments
When evaluating new technologies, processes, or approaches, include systematic assessment of innovation opportunities that might emerge. This involves considering not just whether the primary objective will be achieved, but what secondary benefits or unexpected capabilities might develop during implementation.
Organizational Considerations
Leadership Commitment and Cultural Change
Successful integration of opportunity-based risk management requires genuine leadership commitment to cultural change. Leaders must model behavior that values both threat mitigation and opportunity creation. This means celebrating teams that identify valuable opportunities alongside those that prevent significant risks.
Leadership should establish clear expectations that risk management includes opportunity identification as a core responsibility. Performance metrics, recognition programs, and resource allocation decisions should reflect this balanced approach to uncertainty management.
Training and Capability Development
Teams need specific training to develop opportunity identification skills. While threat identification often comes naturally in quality-conscious cultures, opportunity recognition requires different cognitive approaches and tools.
Training programs should include:
- Techniques for identifying positive potential outcomes
- Methods for evaluating opportunity significance and likelihood
- Approaches for designing controls that enhance opportunities while mitigating risks
- Communication skills for discussing opportunities without compromising analytical rigor
Cross-Functional Integration
Opportunity-based risk management is most effective when integrated across organizational functions. Quality teams might identify process improvement opportunities, while commercial teams recognize market advantages, and technical teams discover innovation possibilities.
Establishing cross-functional opportunity review processes ensures that identified opportunities receive appropriate evaluation and resource allocation regardless of their origin. Regular communication between functions helps build organizational capability to recognize and act on opportunities systematically.
Measuring Success in Opportunity-Based Risk Management
Existing risk management metrics typically focus on negative outcome prevention: deviation rates, incident frequency, compliance scores, and similar measures. While these remain important, opportunity-based programs should also track positive outcome realization.
Enhanced metrics might include:
- Number of opportunities identified per risk assessment
- Percentage of identified opportunities that are successfully realized
- Value generated from opportunity realization (cost savings, quality improvements, efficiency gains)
- Time from opportunity identification to realization
Innovation and Improvement Indicators
Opportunity-focused risk management should drive increased innovation and continuous improvement. Tracking metrics related to process improvements, technology adoption, and innovation initiatives provides insight into the program’s effectiveness in creating value beyond compliance.
Consider monitoring:
- Rate of process improvement implementation
- Success rate of new technology adoptions
- Number of best practices developed and shared across the organization
- Frequency of positive deviations that lead to process optimization
Cultural and Behavioral Measures
The ultimate success of opportunity-based risk management depends on cultural integration. Measuring changes in organizational attitudes, behaviors, and capabilities provides insight into program sustainability and long-term impact.
Relevant measures include:
- Employee engagement with risk management processes
- Frequency of voluntary opportunity reporting
- Cross-functional collaboration on risk and opportunity initiatives
- Leadership participation in opportunity evaluation and resource allocation
Regulatory Considerations and Compliance Integration
Maintaining ICH Q9 Compliance
The opportunity-enhanced approach must maintain full compliance with ICH Q9 requirements while adding value through expanded scope. This means ensuring that all required elements of risk assessment, control, communication, and review continue to receive appropriate attention and documentation.
Regulatory submissions should clearly demonstrate that opportunity identification enhances rather than compromises systematic risk evaluation. Documentation should show how opportunity assessment strengthens process understanding and control strategy development.
Communicating Value to Regulators
Regulators are increasingly interested in risk-based approaches that demonstrate genuine process understanding and continuous improvement capabilities. Opportunity-based risk management can strengthen regulatory relationships by demonstrating sophisticated thinking about process optimization and quality enhancement.
When communicating with regulatory agencies, emphasize how opportunity identification improves process understanding, enhances control strategy development, and supports continuous improvement objectives. Show how the approach leads to better risk control through deeper process knowledge and more robust quality systems.
Global Harmonization Considerations
Different regulatory regions may have varying levels of comfort with opportunity-focused risk management discussions. While the underlying risk management activities remain consistent with global standards, communication approaches should be tailored to regional expectations and preferences.
Focus regulatory communications on how enhanced risk understanding leads to better patient protection and product quality, rather than on business benefits that might appear secondary to regulatory objectives.
Conclusion
Integrating ISO 31000’s opportunity perspective with ICH Q9 compliance represents more than a process enhancement and is a shift toward strategic risk management that positions quality organizations as value creators rather than cost centers. By systematically identifying and capitalizing on positive uncertainties, we can transform quality risk management from a defensive necessity into an offensive capability that drives innovation, efficiency, and competitive advantage.
The framework outlined here provides a practical path forward that maintains regulatory compliance while unlocking the strategic value inherent in comprehensive risk thinking. Success requires leadership commitment, cultural change, and systematic implementation, but the potential returns—in terms of operational excellence, innovation capability, and competitive position—justify the investment.
As we continue to navigate an increasingly complex and uncertain business environment, organizations that master the art of turning uncertainty into opportunity will be best positioned to thrive. The integration of ISO 31000’s risk-as-opportunities approach with ICH Q9 compliance provides a roadmap for achieving this mastery while maintaining the rigorous standards our industry demands.