The United States Pharmacopeia (USP) General Chapter <665> focuses on the plastic components and systems used in the manufacturing of pharmaceutical drug products and biopharmaceutical drug substances and products and is one the fundamental documents for manufacturers, especially in the age of single use.
Key Changes in USP <665>
1. Effective Date Extension
The official effective date for USP <665> has been extended to May 1, 2026. This extension is intended to give stakeholders enough time to follow the new requirements and to align with the development of the ICH Q3E guideline for extractables and leachables (E&Ls).
2. Mandatory Compliance
Previously, USP <665> was informational and not mandatory for compendial compliance. The revisions will make it a mandatory chapter, meaning that compliance will be required for regulatory purposes.
3. Risk-Based Assessments
The revised chapter emphasizes risk-based assessments for the qualification of plastic components. This approach aligns with modern regulatory expectations and provides a structured methodology for evaluating the safety and compatibility of materials used in pharmaceutical manufacturing.
4. Scope and Applicability
USP <665> will cover single-use systems (SUS) and container closure systems used in the storage and processing of pharmaceutical and biopharmaceutical products. This includes guidance on the characterization and qualification of these systems to ensure they do not adversely affect product quality.
5. No Grandfathering of Existing Products
The revisions explicitly state that there will be no grandfathering of existing products. All products, including those already on the market, will need to comply with the new requirements by the effective date.
6. Alignment with USP <1665>
USP <665> is closely related to USP <1665>, which provides guidance on the characterization and qualification processes. Together, these chapters offer a comprehensive framework for managing the risks associated with plastic components in pharmaceutical manufacturing.
7. Stakeholder Engagement
The USP has planned to engage with stakeholders throughout the revision process to ensure that the new requirements are practical and achievable. This includes public notices, comment periods, and collaboration with industry experts.
Implications for Manufacturers
Preparation and Compliance
Manufacturers are advised to start preparing for the changes now to avoid potential backlogs and delays. Early compliance will help make sure a smooth transition and keep product quality.
Global Considerations
While the USP is a U.S.-based standard, its guidelines are often adopted globally. Manufacturers should consider the potential for international regulatory bodies to require equivalency to USP <665> compliance in the future.
Follow a systematic process to validate a shipping container by involving the traditional three main stages: Design Qualification (DQ), Operational Qualification (OQ), and Performance Qualification (PQ).
Design Qualification (DQ)
The DQ stage involves establishing that the shipping container design meets the user requirements and regulatory standards. Key steps include:
Define user requirement specifications (URS) for the container, including temperature range, duration of transport, and product-specific needs.
Review the container design specifications provided by the manufacturer.
Assess the container’s compatibility with the pharmaceutical product and its storage requirements.
Evaluate the container’s compliance with relevant regulatory guidelines and standards.
Operational Qualification (OQ)
OQ involves testing the container under controlled conditions to ensure it operates as intended. This stage includes:
Conducting empty container tests to verify basic functionality.
Testing temperature control systems and monitoring devices.
Evaluating the container’s ability to maintain required conditions under various environmental scenarios.
Assessing the ease of use and any potential operational issues.
Performance Qualification (PQ)
PQ is the most critical stage, involving real-world testing to ensure the container performs as required under actual shipping conditions. Steps include:
Develop a detailed PQ protocol that outlines test conditions, acceptance criteria, and data collection methods.
Conduct shipping trials using actual or simulated product loads.
Test the container under worst-case scenarios, including extreme temperature conditions and extended shipping durations.
Monitor and record temperature data throughout the shipping process.
Assess the impact of various handling conditions (e.g., vibration, shock) on container performance.
Evaluate the container’s performance across different shipping lanes and modes of transport.
Additional Considerations
Associated Materials and Equipment: Ensure all associated materials (e.g., coolants, packaging materials) and monitoring equipment are also qualified.
Re-qualification: For reusable containers, establish a process for periodic re-qualification to ensure ongoing performance.
Documentation: Maintain comprehensive documentation of all qualification stages, including test results, data analysis, and conclusions.
Risk Assessment: Conduct a risk assessment to identify potential failure modes and mitigation strategies.
Best Practices
Use a risk-based approach to determine the extent of testing required for each container type and shipping scenario.
Consider seasonal variations in ambient temperature profiles when designing qualification studies.
Utilize pre-qualified containers from reputable suppliers when possible to streamline the validation process.
Implement a robust change control process to manage any container or shipping process modifications post-validation.
Regularly review and update validation documentation to reflect any changes in regulatory requirements or shipping conditions.
Following this comprehensive approach, you can ensure that your shipping containers are properly validated for pharmaceutical transport, maintaining product quality and integrity throughout the supply chain. Validation is an ongoing process, and containers should be periodically reassessed to ensure continued compliance and performance.
An effective program for managing extractables and leachables (E&L) in biotech involves a comprehensive approach that ensures product safety and compliance with regulatory standards. As single-use technologies have become more prevalent in biopharmaceutical manufacturing, leachables from bags, tubing, and other plastic components have become an area of concern. This has led to more rigorous supplier qualification and leachables risk assessment for single-use systems.
Extractables are chemical compounds that can be extracted from materials (like single-use systems, packaging, or manufacturing equipment) under exaggerated conditions such as elevated temperature, extended contact time, or use of strong solvents. They represent a “worst-case” scenario of chemicals potentially migrating into a drug product. Extractables are specific to the tested material and are independent of the drug product.
Leachables are chemical compounds that actually migrate from materials into the drug product under normal conditions of use or storage. They are specific to the combination of the material and the particular drug substance or product, representing the contaminants that may be present in the final drug formulation. Leachables are typically a subset of extractables that migrate under real-world conditions.
The accumulation of extractables and leachabes in a process fluid is governed by thermodynamics (the extent to which the materials would migrate) and kinetic (the rate at which would migrate) factors, as well as the amount of time during which such migration will occur. Higher temperatures increase the migration rate of leachables from the bulk of plastic to the surface in contact with the process stream or formulation.
Key points
Extractables studies are performed on materials using exaggerated conditions.
Leachables studies are performed on the actual drug product under normal conditions.
Extractables represent potential contaminants, while leachables are actual contaminants.
Both are critical for assessing product safety and quality in biotech manufacturing.
Proper evaluation of extractables and leachables is essential for regulatory compliance and ensuring patient safety in biopharmaceutical products.
Program Objectives
Safety Assurance: Ensure that any chemicals leached from materials into the product do not pose a risk to patient safety.
Regulatory Compliance: Meet all relevant regulatory requirements and guidelines.
Quality Control: Maintain the integrity and quality of the biopharmaceutical product.
Regulatory Requirements
Compliance with USP <661> Plastic Packaging Systems and Their Materials of Construction, and USP <381> Elastomeric Closure for Injection
Compliance with USP <87> Biological Reactivity, In Vitro and USP <88> Biological Reactivity, In Vivo
Compliance with European Pharmacopoeia (EP) requirements for materials used in containers, including EP General Chapter 3.1 Materials Used for the Manufacture of Containers and EP 3.2.9 Rubber Closures
Compliance with Japanese Pharmacopoeia (JP) chapter 7.03 Test for Rubber Closures for Aqueous Infusions
Compliance with EU Commission Decision 97/534/EC for Animal derived stearates
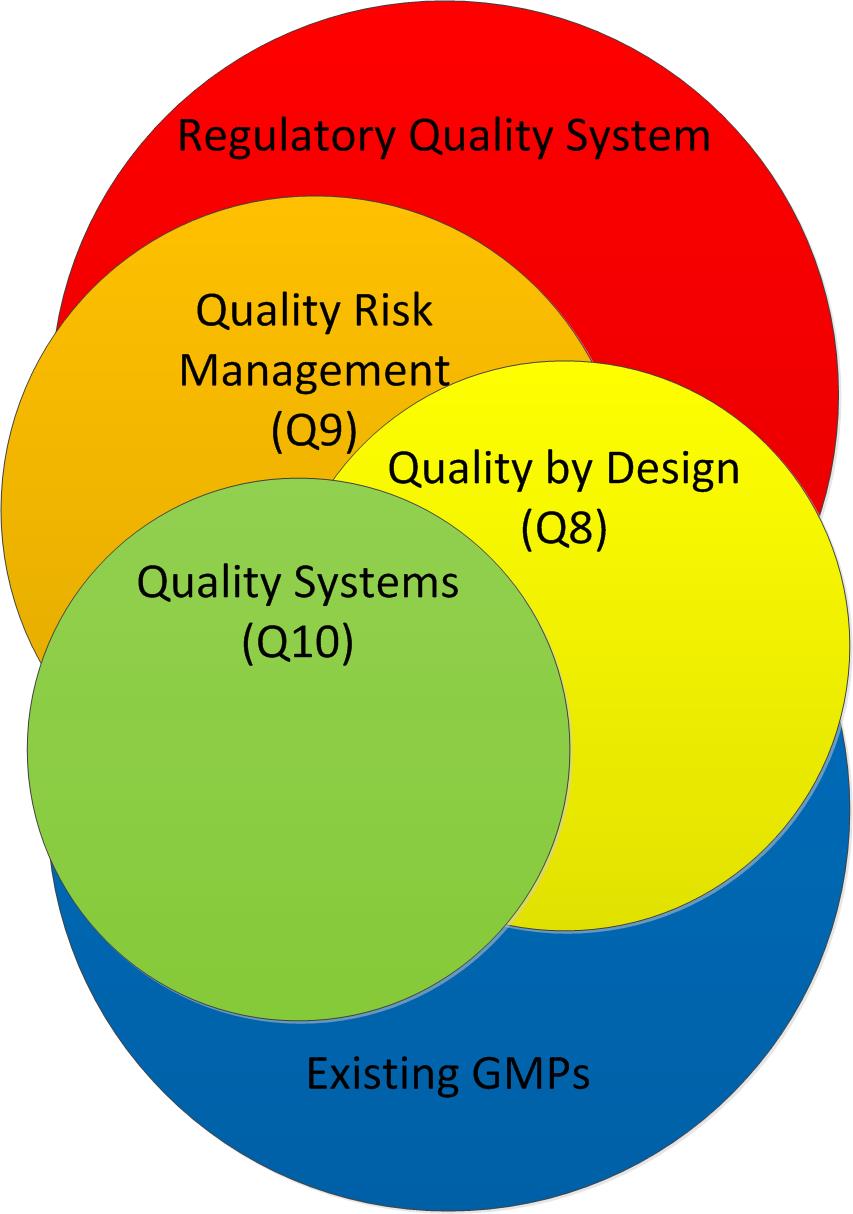
Adherence to ICH Q8, Q9, and Q10 guidelines for quality risk management
Leverage ISO 10993-1:2018 Biological evaluation of medical devices
Program Components
Design Space
The starting point should be a review of the supplier’s data. These studies should be performed on materials at the component level under standardized conditions of temperature time, surface, area, etc., so that the data is representative of intended use, including sterilization techniques. Using this data, the end-user can calculate the minimum amount of extractables based on surface area and other conditions. Consider the impact of dilution and clearance over the complete process through risk assessment and then complement with targeted studies.
These studies should be developed based on Quality-by-design principles described in ICH Q8 to gather all the attributes and parameters used to determine a design space. Scientific variables should be identified to set up the Design of Experiment (DoE) for the testing plan.
Risk Assessment
Material Selection: Evaluate materials used for their potential to release harmful substances.
Process Understanding: Understand the process conditions (e.g., temperature, pH, solvents) that might affect the leaching of chemicals.
Risk Prioritization: Prioritize materials and processes based on their risk of contributing harmful leachables. Consider factors like stage of manufacturing, contact time, and proximity to final product.
The risk assessment needs to be within the overall context of process performance and product safety and efficacy. It should not be a separate risk assessment. You will dive deeper with more specific risk questions, but the hazard identification starts at the process level. In evaluating risks the following factors should be considered:
Proximity of the process steps undergoing a change to the final product. Polymeric components in process steps closer to DS or DP will carry a higher risk rating than those in upstream process steps. For example, a bag or filter used for the final filtration of bulk drug substance (BDS) will have a much higher risk rating than components used in upstream process steps since there are no purification steps post-UF/DF.
Storage and processing conditions (e.g., duration of exposure, temperature, pressure, pH extremes, buffer extraction propensity)
The type of process fluid (e.g., purification buffer versus formulated drug substance, presence of solubilizing agents)
Construction materials
Potential adverse events, including synergistic and additive affects
Drug dose, mode, and frequency of administration
Therapeutic necessity
Your risk assessment will drive study design and should consider:
Analytical challenges
Detecting and quantifying trace levels of leachables, which are often present at extremely low concentrations
Developing analytical methods capable of detecting and quantifying a wide range of potential extractables/leachables
Interference from formulation components or degradation products
Determining appropriate extraction conditions:
Selecting solvents and conditions that adequately simulate or exaggerate real-world use conditions
Balancing the need for aggressive extraction (to identify potential leachables) with realistic use conditions
Toxicological assessment
Evaluating the safety impact of identified extractables/leachables, especially for novel compounds
Determining appropriate safety thresholds and analytical evaluation thresholds
Regulatory expectations
Meeting evolving regulatory requirements and expectations, which can vary between regions
Justifying the extent of E&L studies performed based on risk assessment
Unexpected interactions
Leachables causing unexpected effects, such as oxidation of preservatives or formation of protein-leachable adducts
Interactions between leachables and the drug product that were not predicted by extractables studies
Time and resource constraints
E&L studies can be time-consuming and resource-intensive, potentially impacting development timelines
Absorption issues
Adsorption or absorption of drug product components by single-use materials, potentially affecting product stability or efficacy
Stability considerations
Leachables appearing during stability studies that were not identified in initial extractables screening
Changes in leachables profile over time or under different storage conditions
Material variability
Inconsistencies in extractables/leachables profiles between different lots of materials or components
Biopharmaceutical-specific challenges
Potential impact of leachables on sensitive cell lines or biological processes
Interference of leachables with bioassays or analytical methods specific to biologics
Extractables Studies
Objective: Identify potential extractables from materials under exaggerated conditions.
Methodology:
Use a range of solvents that mimic the process fluids.
Apply exaggerated conditions such as elevated temperatures and extended contact times.
Analyze the extracts using techniques like GC-MS, LC-MS, and ICP-MS.
Data Review: Compare supplier-provided extractable data with the intended use to determine the need for specific studies.
Leachables Studies
Objective: Identify and quantify leachables under actual process conditions.
Methodology:
Conduct studies during the development stages and monitor during stability studies.
Use appropriate solvent systems and conditions that mimic the actual process.
Analyze the product for leachables using validated analytical methods.
Toxicological Assessment: Assess the toxicological impact of identified leachables to ensure they are within safe limits.
Migration Studies
Objective: Evaluate the migration of chemicals from materials into the product over time.
Methodology:
Perform studies during the development phase.
Monitor leachables during formal stability studies under normal and accelerated conditions.
Absorption Studies
Objective: Assess the potential for adsorption or absorption of product components.
Methodology:
Conduct studies if stability issues are observed during hold time studies.
Evaluate the impact on product stability and quality.
Stability Studies
Objective: Ensure the stability of the product in contact with materials.
Methodology:
Conduct real-time and accelerated stability studies.
Monitor product quality attributes such as potency, purity, and safety.
Implementation and Validation
Supplier Qualification
Supplier Evaluation: Assess suppliers’ ability to provide materials that meet E&L requirements.
Documentation Review: Ensure suppliers provide comprehensive extractables data and compliance certificates.
In-House Testing
Validation: Validate the findings from supplier data with in-house testing.
Protocol Development: Develop protocols for E&L testing specific to the product and process conditions.
Acceptance Criteria: Establish acceptance criteria based on regulatory guidelines and risk assessments.
Toxicological Assessment and Risk Mitigation
Assess the toxicological impact of identified leachables to ensure they are within safe limits. Perform Risk Mitigation to:
Implement appropriate controls based on risk assessment results
Consider factors like materials selection, process parameters, and analytical testing
Develop strategies to minimize leachables impact on product quality and safety
Continuous Monitoring
Routine Testing: Implement routine testing of leachables during production.
Change Management: Re-evaluate E&L profiles when there are changes in materials, suppliers, or processes.
Training and Education
Staff Training
Awareness: Train staff on the importance of E&L studies and their impact on product safety.
Technical Training: Provide technical training on conducting E&L studies and interpreting results.
Supplier Collaboration
Engagement: Work closely with suppliers to ensure they understand and meet E&L requirements.
Feedback: Provide feedback to suppliers based on study results to improve material quality.
Conclusion
A robust E&L program in biotech is essential for ensuring product safety, regulatory compliance, and maintaining high-quality standards. By implementing a comprehensive approach that includes risk assessment, thorough testing, supplier qualification, continuous monitoring, and staff training, biotech companies can effectively manage the risks associated with extractables and leachables.
I’m reviewing the status of cleaning validation. Here is the list I’m currently going through, just in case it helps others.
Develop a comprehensive cleaning validation master plan that outlines your overall approach, policies, and procedures for cleaning validation at your facility. This should cover all aspects of the cleaning validation lifecycle.
Ensure you have written standard operating procedures (SOPs) for equipment cleaning processes that address different scenarios (e.g., cleaning between batches, between product changes, etc.).
Have written cleaning validation protocols for each piece of equipment that cover common issues like sampling procedures and analytical methods.
Maintain thorough documentation of your cleaning validation studies, including the protocols, results, and final reports stating whether the cleaning process for each piece of equipment is valid.
Implement a continuous verification program for routine residue monitoring after initial cleaning validation.
Be prepared to demonstrate that your cleaning procedures can consistently clean equipment to predetermined standards using scientifically sound sampling and analytical test methods.
Have data available to support your rationale for residue limits, which should be logical, practical, achievable, and verifiable.
Be ready to explain your approach for different types of equipment (dedicated vs. multi-use) and how you handle potent compounds or other high-risk materials.
Review your cleaning agent selection process and be able to justify the cleaning methods and agents used.
Ensure you have a system in place for equipment maintenance and cleaning records.
Be prepared to discuss how you handle manual vs. automated cleaning processes and any associated validation differences.
Review past audits or inspections and ensure any previous findings related to cleaning validation have been addressed.
A little caveat: I really burnt out on professional obligations last year and have just started to peak my head out. So, it may be a little harder to turn this mad scientist dream into a reality. However, I think it is worth putting out as a thought experiment.
Theme and Scope
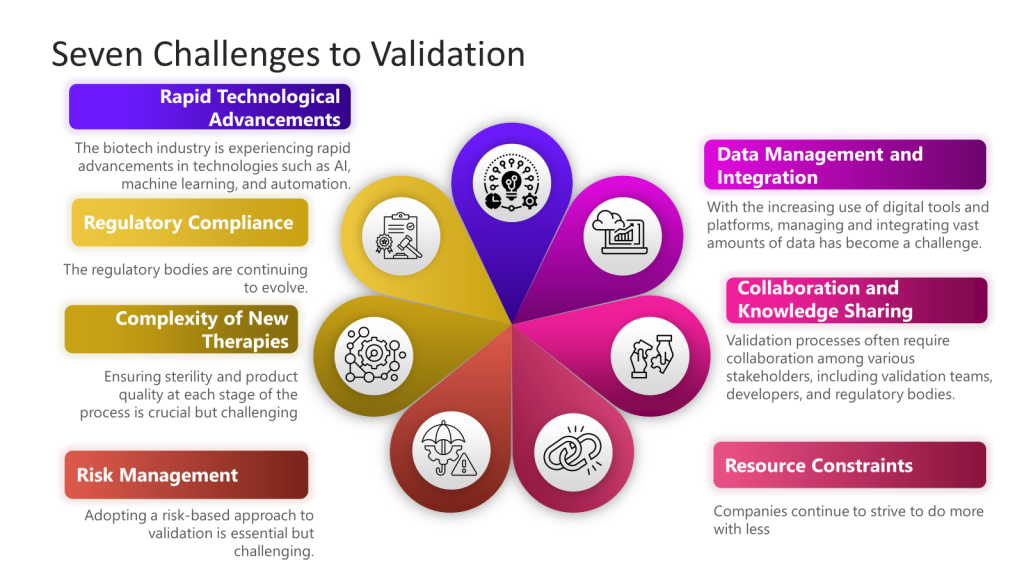
I’ve written a bit about the challenges to quality, and these challenges provide a framework for much of what I think and write about.
More specifically drawing from the “Challenges in Validation” focusing on the challenges of navigating a complex validation landscape characterized by rapid technological advancements, evolving regulatory standards, and the development of novel therapies.
This event would ask, “How do we rise to the challenges of validation in the next decade, leveraging technology and a risk management approach and drawing from the best practices of ASTM E2500, GAMP5, and others to meet and exceed changing regulatory requirements.”
Intended Audience
I go to events, and there are a lot of quality people, OR risk management people, OR computer systems (IT and Q) people, OR engineers, OR analytical method folks, OR process development people. Rarely do I see an event that looks at the whole picture. And rarely do I get to attend an event where we are sharing and blurring the lines between the various silos. So let us break down the silos and invite quality, IT, engineers, and process development individuals involved in the full spectrum of pharmaceutical (and possibly medtech) validation.
This holistic event is meant to blend boundaries, share best practices, challenge ourselves, and look across the entire validation lifecycle.
Structure
Opening/Networking (1 hour)
As people arrive, they go right into a poster event. These posters are each for a specific methodology/approach of ASTM E2500, ISPE Baseline Guides, FDA’s Guidance for Process Validation: General Principles and Practices, ICH’s QbD approach, and GAMP5. Maybe some other things.
These posters would each:
Provide an overview of what it is and why it is important
Overview of methodology
What challenges it overcomes
Lessons that can be applied
Challenges/problems inherent in the approach
These posters would be fun to develop and take a good squad of experts.
After an hour of mingling, sharing, and baselining, we could move to the next step.
Fish Bowl Debate (45 minutes)
Having earlier selected a specific topic and a panel of experts, hold a fish bowl debate. This would be excellent as a mock-inspection, maybe of a really challenging topic. Great place to bring those inspectors in.
During a fish bowl, everyone not in the center is taking notes. I love a worksheet to help with this by providing things to look for to get the critical thinking going.
Future Workshop (1.5 hour)
Introduce the activity (10 min)
Ask participants to reflect on their present-day situation, write down all their negative experiences on sticky notes, and place them on the wall. (15 min)
Invite participants to list uncertainties they face by asking, “In your/our operating environment, what factors are impossible to predict or control their direction?” (5 min).
Prioritize the most critical factors by asking, “Which factors threaten your/our ability to operate successfully?” (10 min)
Based on the group’s history and experience, select the two most critical and most uncertain (X and Y). (5 min)
Create a grid with two axes—X & Y—with a “more of <— —> less of” continuum to represent the factor on each axis. For example, suppose new modalities are a critically uncertain factor for the X-axis. In that case, one end of the X-axis is many new modalities, and the other is no new modalities. Repeat for the Y factor and axis. For instance, if patent protection is a critical factor, one end of the Y axis is strong patent protection, and the other has no patent protection. Four quadrants are created. (5 min)
Break into four groups, and each group creatively names and writes a thumbnail scenario for one of the quadrants. (10 min)
The four groups share their scenarios briefly. 2 min. each
Participants fantasize about the desired future situation. How would the ideal situation be for them? At this stage, there are no limitations; everything is possible. Write on stick notes and apply them to the most likely quadrant. (10 minutes)
Do a n/3 activity to find the top ideas (enough for groups of 4-5 each) (3 min)
Explain the next activity (2 min)
Lunch (1 hour)
Open Space Solution (1 hour)
For each top idea, the participants vote with their feet and go to develop the concept. Each group is looking to come up with the challenge solved, a tool/methodology, and an example.
Review the Results of the Open Space Solutions (1 hour)
Each team presents for 5-8 minutes.
1-2-4-All (20 minutes)
Silent self-reflection by individuals on the shared challenge, framed as a question “What opportunities do YOU see for making progress on this challenge? How would you handle this situation? What ideas or actions do you recommend?” (1 min)
Generate ideas in pairs, building on ideas from self-reflection. (2 min)
Share and develop ideas from your pair in foursomes (notice similarities and differences).( 4 min)
Ask, “What is one idea that stood out in your conversation?” Each group shares one important idea with all (15 min)
Closing Commitment (5 min)
Where will this live? What comes next? Make a commitment to follow up electronically.
Networking
Spend an hour or so with drinks and food and discuss everything. Never enough socialization.




{kind=link}