I don’t like the term validation deviation, preferring to use discrepancy to cover the errors or failures that occur during qualification/validation, such as when the actual results of a test step in a protocol do not match the expected results. These discrepancies can arise for various reasons, including errors in the protocol, execution issues, or external factors.
I don’t like using the term deviation as I try to avoid terms becoming too overused in too many ways. By choosing discrepancy it serves to move them to a lower order of problem so they can be addressed holistically.
Validation discrepancies really get to the heart of deciding whether the given system/process is fit-for-purpose and fit-for-use. As such, they require being addressed in a timely and pragmatic way.
And, like anything else, having an effective procedure to manage is critical.
Validation discrepancies are a great example of building problem-solving into a process.
In ASTM E2500, a Subject Matter Expert (SME) is an individual with specialized knowledge and technical understanding of critical aspects of manufacturing systems and equipment. The SME plays a crucial role throughout the project lifecycle, from defining needs to verifying and accepting systems. They are responsible for identifying critical aspects, reviewing system designs, developing verification strategies, and leading quality risk management efforts. SMEs ensure manufacturing systems are designed and verified to meet product quality and patient safety requirements.
In the ASTM E2500 process, the Subject Matter Experts (SME) has several key responsibilities critical to successfully implementing the standard. These responsibilities include:
Definition of Needs: SMEs define the system’s needs and identify critical aspects that impact product quality and patient safety.
Risk Management: SMEs participate in risk management activities, helping to identify, assess, and manage risks throughout the project lifecycle. This includes conducting quality risk analyses and consistently applying risk management principles.
Verification Strategy Development: SMEs are responsible for planning and defining verification strategies. This involves selecting appropriate test methods, defining acceptance criteria, and ensuring that verification activities are aligned with the project’s critical aspects.
System Design Review: SMEs review system designs to ensure they meet specified requirements and address identified risks. This includes participating in design reviews and providing technical input to optimize system functionality and compliance.
Execution of Verification Tests: SMEs lead the execution of verification tests, ensuring that tests are conducted accurately and that results are thoroughly reviewed. They may also leverage vendor documentation and test results as part of the verification process, provided the vendor’s quality system and technical capabilities are deemed acceptable.
Change Management: SMEs play a crucial role in change management, ensuring that any modifications to the system are properly evaluated, documented, and implemented. This helps maintain the system’s validated state and ensures continuous compliance with regulatory requirements.
Continuous Improvement: SMEs are involved in continuous process improvement efforts, using operational and performance data to identify opportunities for enhancements. They also conduct root-cause analyses of failures and implement technically sound improvements based on gained product knowledge and understanding.
These responsibilities highlight the SME’s integral role in ensuring that manufacturing systems are designed, verified, and maintained to meet the highest standards of quality and safety, as outlined in ASTM E2500.
The ASTM E2500 SME is a Process Owner
ASTM E2500 uses the term SME in the same way we discuss process owners, or what is sometimes called product or molecule stewards. The term should probably be changed to reflect the special role of the SME and the relationship with other stakeholders.
A Molecule Steward has a specialized role within pharmaceutical and biotechnology companies and oversees the lifecycle of a specific molecule or drug product. This role involves a range of responsibilities, including:
Technical Expertise: Acting as the subject matter expert per ASTM E2500.
Product Control Strategies: Implementing appropriate product control strategies across development and manufacturing sites based on anticipated needs.
Lifecycle Management: Providing end-to-end accountability for a given molecule, from development to late-stage lifecycle management.
A Molecule Steward ensures a drug product’s successful development, manufacturing, and lifecycle management, maintaining high standards of quality and compliance throughout the process.
The ASTM E2500 SME (Molecule Steward) and Stakeholders
In the ASTM E2500 approach, the Subject Matter Expert (Molecule Steward) collaborates closely with various project players to ensure the successful implementation of manufacturing systems.
Definition of Needs and Requirements
Collaboration with Project Teams: SMEs work with project teams from the beginning to define the system’s needs and requirements. This involves identifying critical aspects that impact product quality and patient safety.
Input from Multiple Departments: SMEs gather input from different departments, including product/process development, engineering, automation, and validation, to ensure that all critical quality attributes (CQAs) and critical process parameters (CPPs) are considered.
Risk Management
Quality Risk Analysis: SMEs lead the quality risk analysis process, collaborating with QA and other stakeholders to identify and assess risks. This helps focus on critical aspects and consistently apply risk management principles.
Vendor Collaboration: SMEs often work with vendors to leverage their expertise in conducting risk assessments and ensuring that vendor documentation meets quality requirements.
System Design Review
Design Review Meetings: SMEs participate in design review meetings with suppliers and project teams to ensure the system design meets the defined needs and critical aspects. This collaborative effort helps in reducing the need for modifications and repeat tests.
Supplier Engagement: SMEs engage with suppliers to ensure their design solutions are understood and integrated into the project. This includes reviewing supplier documentation and ensuring compliance with regulatory requirements.
Verification Strategy Development
Developing Verification Plans: SMEs collaborate with QA and engineering teams to develop verification strategies and plans. This involves selecting appropriate test methods, defining acceptance criteria, and ensuring verification activities align with project goals.
Execution of Verification Tests: SMEs may work with suppliers to conduct verification tests at the supplier’s site, ensuring that tests are performed accurately and efficiently. This collaboration helps achieve the “right test” at the “right time” objective.
Change Management
Managing Changes: SMEs play a crucial role in the change management process, working with project teams to evaluate, document, and implement changes. This ensures that the system remains in a validated state and continues to meet regulatory requirements.
Continuous Improvement: SMEs collaborate with other stakeholders to identify opportunities for process improvements and implement changes based on operational and performance data.
Documentation and Communication
Clear Communication: SMEs ensure clear communication and documentation of all verification activities and acceptance criteria. This involves working closely with QA to validate all critical aspects and ensure compliance with regulatory standards.
A system boundary for equipment or utility refers to the demarcation points that define the extent of a system’s components and the scope of its operations. This boundary is crucial for managing, validating, maintaining, and securing the system.
For utilities, the last valve before the system being supplied can be used as the boundary, which can also serve as a Lock Out Tag Out (LOTO) point.
Physical connections like tri-clamp connections or flanges can define the boundary for packaged systems or skids.
For critical and non-critical systems, such as air or HVAC systems, filters can be the boundary between systems.
Defining system boundaries is crucial during the design of equipment and systems. It helps identify where the equipment starts and stops and where the breakpoints are situated. This ensures a smooth transition and handover during the commissioning process.
Early Definition: Define system boundaries as early as possible in the system’s development life cycle to reduce costs and ensure effective security controls are implemented from the start.
Stakeholder Involvement: Relevant stakeholders, such as system engineers, utility providers, and maintenance teams, should be involved in defining system boundaries to ensure alignment and a clear understanding of responsibilities.
Documentation and Traceability: To ensure consistency and traceability, document and maintain system boundaries in relevant diagrams (e.g., P&IDs, system architecture diagrams) and commissioning/qualification protocols.
Periodic Review: Regularly review and update system boundaries as the system evolves or the environment changes, using change management and configuration management processes to ensure consistency and completeness.
Enterprise-level Coordination: At an enterprise level, coordinate and align system boundaries across all major systems to identify gaps, overlaps, and seamless coverage of security responsibilities.
Applying Systems Thinking
Systems thinking and modeling techniques are essential for managing and improving complex systems. These approaches help understand the interconnected nature of systems, identify key variables, and make informed decisions to enhance performance, reliability, and sustainability. Here’s how these methodologies can be applied:
Holistic Approach
Systems thinking involves viewing the system as an integrated whole rather than isolated components. This approach acknowledges that the system has qualities that the sum of individual parts cannot explain.
When developing frameworks, models, and best practices for systems, consider the interactions between people, processes, technology, and the environment.
Key Elements:
Interconnectedness: Recognize that all parts of the utility system are interconnected. Changes in one part can affect other parts, sometimes in unexpected ways.
Feedback Loops: Identify feedback loops where outputs from one part of the system influence other parts. These can be reinforcing or balancing loops that affect system behavior over time.
Time Consideration: Understand that effects rarely ripple through a complex system instantaneously. Consider how changes will affect the system over time.
ASTM E2500, the Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment, is intended to “satisfy international regulatory expectations in ensuring that manufacturing systems and equipment are fit for the intended use and to satisfy requirements for design, installation, operation, and performance.”
The ASTM E2500 approach is a comprehensive framework for specification setting, design, and verification of pharmaceutical and biopharmaceutical manufacturing systems and equipment. It emphasizes a risk- and science-based methodology to ensure that systems are fit for their intended use, ultimately aiming to enhance product quality and patient safety.
Despite its 17-year history, it is fair to say it is not the best-implemented standard. There are still many unrealized opportunities and some major challenges. I don’t think a single organization I’ve been in has fully aligned, and ASTM E2500 can feel aspirational.
Key Principles
Risk Management: The approach integrates risk management principles from ICH Q8, Q9, and Q10, focusing on identifying and mitigating risks to product quality and patient safety throughout the lifecycle of the manufacturing system.
Good Engineering Practices (GEP): It incorporates GEP to ensure systems are correctly designed, installed, and operated.
Flexibility and Efficiency: It strives for a more flexible and efficient organization of verification activities that can be adapted to each project’s specific context.
Regulatory agencies expect drugmakers to persuade them that we know our processes and that our facilities, equipment, systems, utilities, and procedures have been established based on concrete data and a thorough risk assessment. The ASTM E2500 standard provides a means of demonstrating that all of these factors have been validated in consideration of carefully evaluated risks.
What the Standard Calls for
Four Main Steps
Requirements: Define the system’s needs and critical aspects. Subject Matter Experts (SMEs) play a crucial role in this phase by defining needs, identifying critical aspects, and developing the verification strategy.
Specification & Design: Develop detailed specifications and design the system to meet the requirements. This step involves thorough design reviews and risk assessments to ensure the system functions as intended.
Verification: Conduct verification activities to confirm that the system meets all specified requirements. This step replaces the traditional FAT/SAT/IQ/OQ/PQ sequence with a more streamlined verification process that can be tailored to the project’s needs.
Acceptance & Release: Finalize the verification process and release the system for operational use. This step includes the final review and approval of all verification activities and documentation.
Four Cross-Functional Processes
Good Engineering Practices (GEP): Ensure all engineering activities adhere to industry standards and best practices.
Quality Risk Management: Continuously assess and manage risks to product quality and patient safety throughout the project.
Design Review: Regularly reviews the system design to ensure it meets all requirements and addresses identified risks.
Change Management: Implement a structured process for managing system changes to ensure that all modifications are appropriately evaluated and documented.
Applications and Benefits
Applicability: The ASTM E2500 approach can be applied to new and existing manufacturing systems, including laboratory, information, and medical device manufacturing systems.
Lifecycle Coverage: It applies throughout the manufacturing system’s lifecycle, from concept to retirement.
Regulatory Compliance: The approach is designed to conform with FDA, EU, and other international regulations, ensuring that systems are qualified and meet all regulatory expectations.
Efficiency and Cost Management: By focusing on critical aspects and leveraging risk management tools, the ASTM E2500 approach can streamline project execution, reduce time to market, and optimize resource utilization.
The ASTM E2500 approach provides a structured, risk-based framework for specifying, designing, and verifying pharmaceutical and biopharmaceutical manufacturing systems. It emphasizes flexibility, efficiency, and regulatory compliance, making it a valuable tool for ensuring product quality and patient safety.
What Makes it Different?
ASTM E2500
The more traditional approach
Testing Approach
It emphasizes a risk-based approach, focusing on identifying and managing risks to product quality and patient safety throughout the manufacturing system’s lifecycle. This approach allows for flexibility in organizing verification activities based on the specific context and critical aspects of the system.
Typically follows a prescriptive sequence of tests (FAT, SAT, IQ, OQ, PQ) as outlined in guidelines like EU GMP Annex 15. This method is more rigid and less adaptable to the specific needs and risks of each project.
Verification vs Qualification
The term “verification” encompasses all testing activities, which can be organized more freely and rationally to optimize efficiency. Verification activities are tailored to the project’s needs and focus on critical aspects.
Follows a structured qualification process (Installation Qualification, Operational Qualification, Performance Qualification) with predefined steps and documentation requirements.
Role of Subject Matter Experts
SMEs play a crucial role from the start of the project, contributing to the definition of needs, identification of critical aspects, system design review, and development of the verification strategy. They are involved throughout the project lifecycle.
SMEs are typically involved at specific points in the project lifecycle, primarily during the qualification phases, and may not have as continuous a role as in the ASTM E2500 approach.
Integration of Good Engineering Practices
Offers greater flexibility in organizing verification activities, allowing for a more efficient and streamlined process. This can lead to reduced time to market and optimized resource utilization.
While GEP is also important, the focus is more on the qualification steps rather than integrating GEP throughout the entire project lifecycle.
Change Management
It emphasizes early and continuous change management, starting from the supplier’s site, to avoid test duplication and ensure that changes are properly evaluated and documented.
It emphasizes early and continuous change management, starting from the supplier’s site, to avoid test duplication and ensure that changes are properly evaluated and documented.
Documentation
Documentation is focused on risk management and verification activities, ensuring compliance with international regulations (FDA, EU, ICH Q8, Q9, Q10). The approach is designed to meet regulatory expectations while allowing for flexibility in documentation.
quires extensive documentation for each qualification step, which can be more cumbersome and less adaptable to specific project needs.
Opinion
I’m watching to see what the upcoming update to Annex 15 will do to address the difficulties some see between an ATSM E2500 approach and the European regulations. I also hope we will see an update to ISPE Baseline® Guide Volume 5: Commissioning and Qualification to align an approach.
ISPE Baseline® Guide Volume 5
ATSM E2500
Design inputs Impact assessment Design Qualification Commissioning Multiple trial runs to get things right IQ, OQ, PQ, and acceptance criteria GEP Scope and QA Scope overlapped Focused on Documentation Deliverables Change Management
Design inputs Design Review Risk Mitigation Critical Control Parameters define Acceptance Criteria Verification Testing Performance Testing GEP Scope and QA Scope have a clear boundary Process, Product Quality and Patient Safety Quality by Design, Design Space, and Continuous Improvement
To be honest I don’t think ATSM E2500, ISPE Guide 5, or anything else has the balance just right. And your program ends up being a triangulation between these and the regulations. And don’t even bring in trying to align GAMP5 or USP <1058> or…or…or…
And yes, I do consider this part of my 3-year plan. I look forward to the challenges of a culture shift, increased SME involvement, formalization of GEPs (and teaching engineers how to write), effective change management, timely risk assessments, and comprehensive implementation planning.
Risk management plays a pivotal role in validation by enabling a risk-based approach to defining validation strategies, ensuring regulatory compliance, mitigating product quality and safety risks, facilitating continuous improvement, and promoting cross-functional collaboration. Integrating risk management principles into the validation lifecycle is essential for maintaining control and consistently producing high-quality products in regulated industries such as biotech and medical devices.
We will conduct various risk assessments in our process lifecycle—many ad hoc (static) and a few living (dynamic). Understanding how they fit together in a larger activity set is crucial.
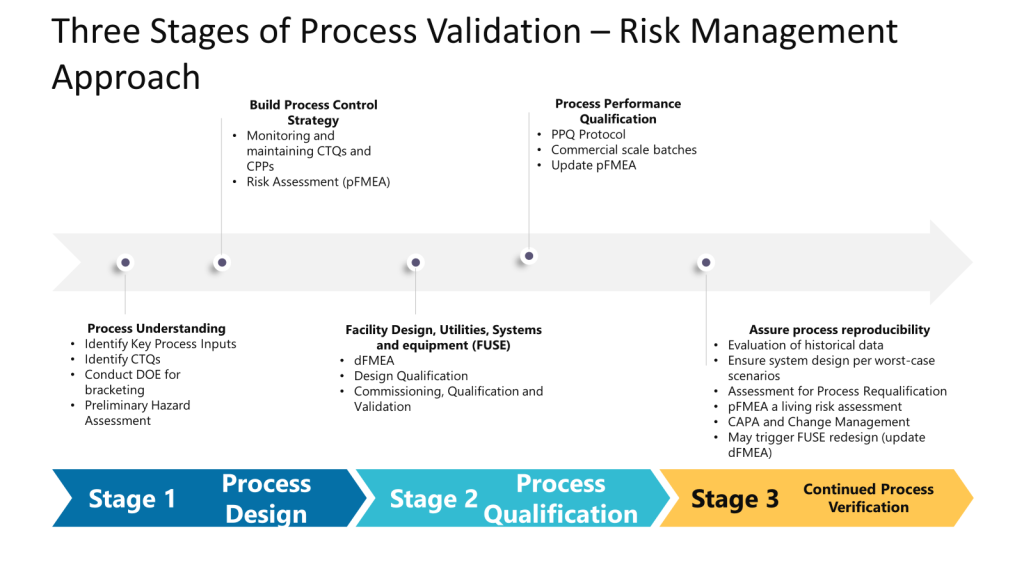
In the Facility, Utilities, Systems, and Equipment (FUSE) space, we are taking the process understanding, translating it into a design, and then performing Design Qualification (DQ) to verify that the critical aspects (CAs) and critical design elements (CDEs) necessary to control risks identified during the quality risk assessment (QRA) are present in the design. This helps mitigate risks to product quality and patient safety. To do this, we need to properly understand the process. Unfortunately, we often start with design before understanding the process and then need to go back and perform rework. Too often I see a dFMEA ignored or as an input to the pFMEA instead of working together in a full risk management cycle.
The Preliminary Hazard Analysis (PHA) supports a pFMEA, which supports a dFMEA, which supports the pFMEA (which also benefits at this stage from a HAACP). Tools fit together to provide the approach. Tools do not become the approach.
Design and Process FMEAs
DFMEA (Design Failure Mode and Effects Analysis) and PFMEA (Process Failure Mode and Effects Analysis) are both methodologies used within the broader FMEA framework to identify and mitigate potential failures. Still, they focus on different aspects of development and manufacturing.
DFMEA
PFMEA
Scope and Focus
Primarily scrutinizes design to preempt flaws.
Focuses on processes to ensure effectiveness, efficiency and reliability.
Stakeholder Involvement
Engages design-oriented teams like engineering, quality engineers, and reliability engineers.
Involves operation-centric personnel such as manufacturing, quality control, quality operations, and process engineers.
Inputs and Outputs
Relies on design requirements, product specs, and component interactions to craft a robust product.
Utilizes process steps, equipment capabilities, and parameters to design a stable operational process.
Stages in lifecycle
Conducted early in development, concurrent with the design phase, it aids in early issue detection and minimizes design impact.
Executed in production planning post-finalized design, ensuring optimized operations prior to full-scale production.
Updated When
Executed in production planning post-finalized design, ensuring optimized operations before full-scale production.
The design qualification phase is especially suitable for determining risks for products and patients stemming from the equipment or machine. These risks should be identified during the design qualification and reflected by appropriate measures in the draft design so that the operator can effectively eliminate, adequately control, and monitor or observe them. To identify design defects (mechanical) or in the creation of systems (electronics) on time and to eliminate them at a low cost, it is advisable to perform the following risk analysis activities for systems, equipment, or processes:
Categorize the GMP criticality and identify the critical quality attributes and process parameters;
Categorize the requirements regarding the patient impact and product impact (for example, in the form of a trace matrix);
Identify critical functions and system elements (e.g., the definition of a calibration concept and preventive maintenance);
Investigate functions for defect recognition. This includes checking alarms and fault indications, operator error, etc. The result of this risk analysis may be the definition of further maintenance activities, a different assessment of a measurement point, or the identification of topics to include in the operating manuals or procedures.
Additional risk analyses for verifying the design may include usability studies using equipment mock-ups or preliminary production trials (engineering studies) regarding selected topics to prove the feasibility of specific design aspects (e.g., interaction between machine and materials).
Too often, we misunderstand risk assessments and start doing them at the most granular level. This approach allows us to right-size our risk assessments and holistically look at the entire lifecycle.