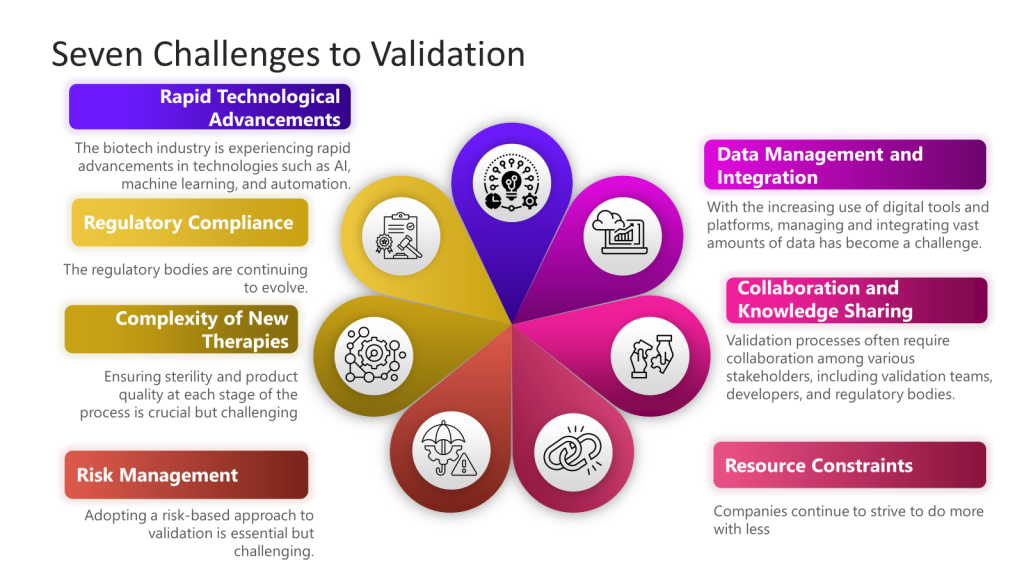
It is crucial for a Marketing Authorization Holder (MAH) to review and approve changes made by a Contract Development and Manufacturing Organization (CDMO) for several important reasons:
Regulatory Compliance
The Market Authorization Holder (MAH) – or the sponsor for pre-commercial GMP manufacturing – bears the primary responsibility for ensuring compliance with the marketing authorization and regulatory requirements throughout the product’s lifecycle. By reviewing and approving CDMO changes, the MAH can:
- Ensure changes align with the approved marketing authorization
- Verify that any variations to the marketing authorization are properly submitted to regulatory authorities
- Maintain oversight of post-approval change management as required by regulations
Before I go any further on the topic I want you to go and read my post Classification of Changes for GMP/GDP. This post will build on that discussion.
I think it is better for the CDMO to put a lot of thought into this, and the MAH (the client) to evaluate and adapt. For all but the big players, the volume is going to be on the CDMO’s side. But if you are the client and your CDMO hasn’t taken this into account to the appropriate degree, you need to ensure appropriate steps taken. As such the rest of this post will be written from the CDMO’s side, but the same principles apply to the MAH (and should be included in the audit program).
Remember we have three goals:
- Fulfill our contractual responsibilities
- Help the MAH maintain appropriate control as the product owner
- Ensure alignment between both parties on change implementation
The critical requirement here is ensuring the right changes get to the right client so they can be filled the right way. Returning to basics, we are approaching changes as:

Now it’s easy to apply this to product. Create and/or receive the design space and the control space. Everything that falls into a non-established condition does not get reported to the client at time of execution. If it is “Do and Report” is is in the APQR. If it is “Do and Record” they can see it during the audit.
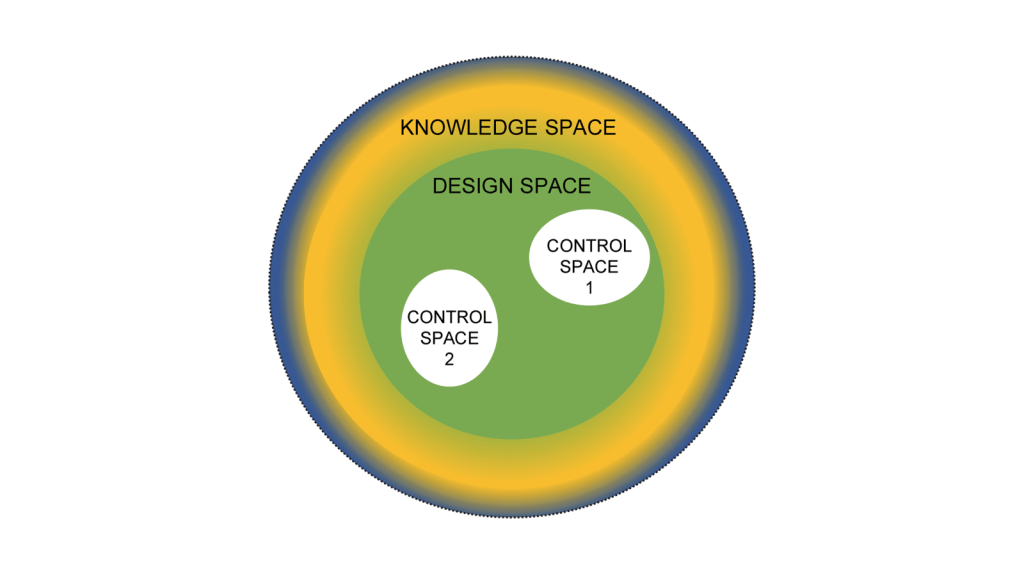
Where a lot of CDMOs trip up here are facility and quality system changes. My recommendation here is the same, define a design space based on the CMC section of the Common Technical Document which basically boils down to:
The CMC (Chemistry, Manufacturing, and Controls) section of a regulatory dossier typically includes the following key facility-related information:
- Manufacturing Facilities
- Names and addresses of all manufacturing, testing, and storage facilities involved in production
- Description of the manufacturing operations performed at each site
- Floor plans and layouts of production areas
- Details on utilities and support systems (HVAC, water, gases, etc.)
- Information on facility design features for contamination control and product protection
- Equipment
- List of major production and laboratory equipment
- Equipment specifications and capacities
- Cleaning and maintenance procedures for equipment
- Environmental Controls
- Description of clean room classifications and environmental monitoring programs
- Air handling systems and controls
- Water systems (purified water, water for injection) and controls
- Material Flow
- Personnel and material flow diagrams
- Segregation of operations to prevent cross-contamination
- Quality Control Laboratories
- Description of QC lab facilities and equipment
- Environmental controls in QC labs
- Storage Areas
- Description of storage facilities for raw materials, intermediates, and finished products
- Storage conditions and controls (temperature, humidity, etc.)
There is a whole lot of wiggle room here in things that fall into “Do and Record.” By building this into your change control system you can delineate what goes to to the client and what doesn’t. I recommend sitting down with this list and deciding what types of changes fall into “Tell and Do” – what you ask permission from clients before doing; “Do and Report” – what goes in the APQR; and, “Do and Record” – what the client sees when they audit.
You know have good rules on what changes go to a client for prior approval and which ones do not. This gets codified in two places: the change control process and the quality/technical agreement.
Some other things to build into your change control process:
- Documenting when a client requests a change, the reason and the impact on the platform. Remember you have other clients, and more and more CDMO’s are offering a platform, so there needs to be appropriate review and endorsement.
- Think through how changes to facility (and other platform elements) are communicated and gated for multiple clients. Have a mechanism to manage client specific activities and to track first-product impacted for multiple products.
- Have clear timelines and expectations on change communication and approval with the client in the quality/technical agreement. Hold each other accountable.
- Have contingency plans. There will always be that one client who will be in shortage if you make that urgent change just when you want/need to.
- Have a method for evaluating requested changes to the change plan by clients and making decisions around it. There will be that one client who doesn’t agree or wants something weird that disagrees with what all the other clients want.
- Have rules in place to manage changes inactive for long periods or extensions specific for those changes that rise to client approval. These will have a different flow than internal changes.
I’ve used a bit of commercial headspace for this post, relying on the APQR. For clinical processes, product tends to fall into campaign-mindset, so “Do and Report” ends up being more a clinical campaign change report than an APQR.