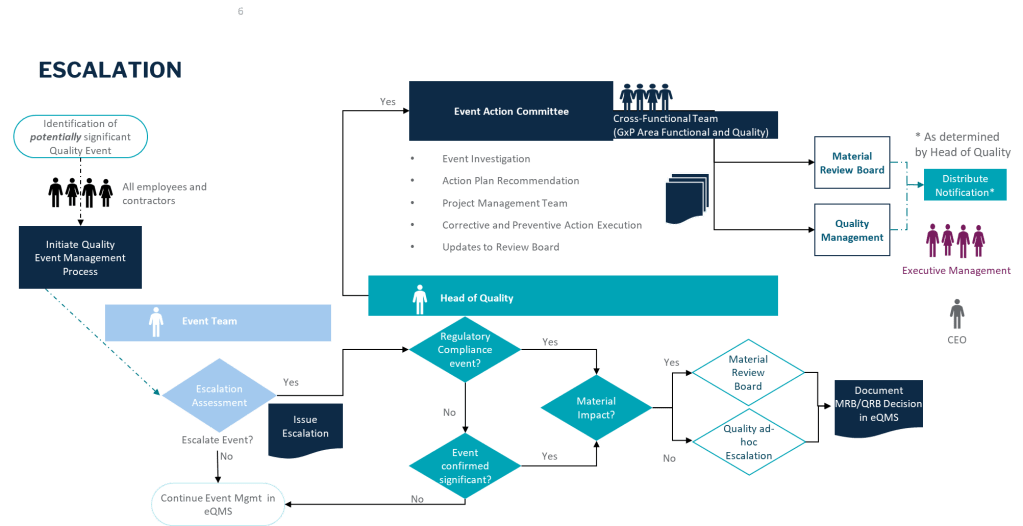
I think it is unfortunate that two of the world’s most influential regulatory agencies, the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA), have taken markedly different approaches to transparency in sharing Good Manufacturing Practice (GMP) observations and non-compliance information with the public.
The Foundation of Regulatory Transparency
FDA’s Transparency Initiative
The FDA’s commitment to transparency traces back to the Freedom of Information Act (FOIA) of 1966, which required federal agencies to provide information to the public upon request. However, the agency’s proactive transparency efforts gained significant momentum under President Obama’s Open Government Initiative. In June 2009, FDA Commissioner Dr. Margaret Hamburg launched the FDA’s Transparency Initiative, creating new webpages, establishing FDA-TRACK performance monitoring system, and proposing steps to provide greater public understanding of FDA decision-making.
EMA’s Evolution Toward Transparency
The EMA’s journey toward transparency has been more gradual and complex For many years, EU inspectorates did not publish results of their inspections, unlike the FDA’s long-standing practice of making Form 483s and Warning Letters publicly accessible. This changed significantly in 2014 when the EMA launched a new version of the EudraGMDP database that included, for the first time, the publication of statements of non-compliance with Good Manufacturing Practice.
The EMA’s approach to transparency reflects its commitment to transparency, efficiency, and public health protection through structured partnerships with agencies worldwide 1. However, the agency’s transparency policy has faced criticism for being “marred by too many failings,” particularly regarding pharmaceutical companies’ ability to redact clinical study reports.
FDA’s Comprehensive Data Infrastructure
The FDA operates several interconnected systems for sharing inspection and compliance information:
Form 483 Database and Public Access
The FDA maintains extensive databases for Form 483 inspectional observations, which are publicly accessible through multiple channels. The agency’s Office of Inspections and Investigations provides spreadsheets summarizing inspection observations by fiscal year, broken down by product areas including biologics, drugs, devices, and other categories.
FDA Data Dashboard
Launched as part of the agency’s transparency initiative, the FDA Data Dashboard presents compliance, inspection, and recall data in an easy-to-read graphical format. The dashboard provides data from FY 2009 onward and allows access to information on inspections, warning letters, seizures, injunctions, and recall statistics. The system is updated semi-annually and allows users to download information, manipulate data views, and export charts for analysis.
Warning Letters and Public Documentation
All FDA-issued Warning Letters are posted on FDA.gov in redacted form to permit public access without requiring formal FOIA requests. This practice has been in place for many years, with warning letters being publicly accessible under the Freedom of Information Act.
- https://www.fda.gov/about-fda/office-inspections-and-investigations/oii-foia-electronic-reading-room
EMA’s EudraGMDP Database
The EMA’s primary transparency tool is the EudraGMDP database, which serves as the Community database on manufacturing, import, and wholesale-distribution authorizations, along with GMP and GDP certificates. A public version of the database has been available since 2011, providing access to information that is not commercially or personally confidential.
The EudraGMDP database contains several modules including Manufacturing Import Authorisation (MIA), GMP certificates, Wholesale Distribution Authorisation (WDA), and Active Product Ingredient Registration (API REG). The database is publicly accessible without login requirements and is maintained by the EMA with data populated by EEA national competent authorities.
Non-Compliance Reporting and Publication
A significant milestone in EMA transparency occurred in 2014 when the agency began publishing statements of non-compliance with GMP . These documents contain information about the nature of non-compliance and actions taken by issuing authorities to protect public health, aiming to establish coordinated responses by EU medicines regulators.
A major difference here is that the EMA removes non-compliance statements from EudraGMDP following successful compliance restoration. The EMA’s procedures explicitly provide for post-publication modifications of non-compliance information. Following publication, the lead inspectorate authority may modify non-compliance information entered in EudraGMDP, for example, following receipt of new information, with modified statements distributed to the rapid alert distribution list.
This is unfortunate, as it requires going to a 3rd party service to find historical data on a site.
| Category | FDA | EMA |
|---|---|---|
| Volume of Published Information | Over 25,000 Form 483s in databases | 83 non-compliance reports total (2007-2020) |
| Annual Inspection Volume | Every 483 observation is trackable at a high level | Limited data available |
| Database Update Frequency | Monthly updates to inspection databases | Updates as available from member states |
| Dashboard Updates | Semi-annual updates | Not applicable |
| Historical Data Availability | Form 483s and warning letters accessible for decades under FOIA | Non-compliance information public since 2014 |
| Information Scope | Inspections, warning letters, seizures, injunctions, recalls, import alerts | Primarily GMP/GDP certificates and non-compliance statements |
| Geographic Distribution of Non-Compliance | Global coverage with detailed breakdowns | India: 35 reports, China: 22 reports, US: 4 reports |
| Real-Time Access | Yes – monthly database updates | Limited – dependent on member state reporting |
| Public Accessibility | Multiple channels: direct database access, FOIA requests | Single portal: EudraGMDP database |
| Data Manipulation Capabilities | Users can download, manipulate data views, export charts | Basic search and view functionality |
| Login Requirements | No login required for public databases | No login required for EudraGMDP |
| Commercial Confidentiality | Redacted information | Commercially confidential information not published |
| Non-Compliance Statement Removal | Form 483s remain public permanently | Statements can be removed after successful remediation |
While both the FDA and EMA have made significant strides in regulatory transparency, the FDA clearly shares more information about GMP observations and non-compliance issues. The FDA’s transparency advantage stems from its longer history of public disclosure under FOIA, more comprehensive database systems, higher volume of published enforcement actions, and more frequent updates to public information.
My next post will be on the recent changes at the FDA and what that means for ongoing transparency.