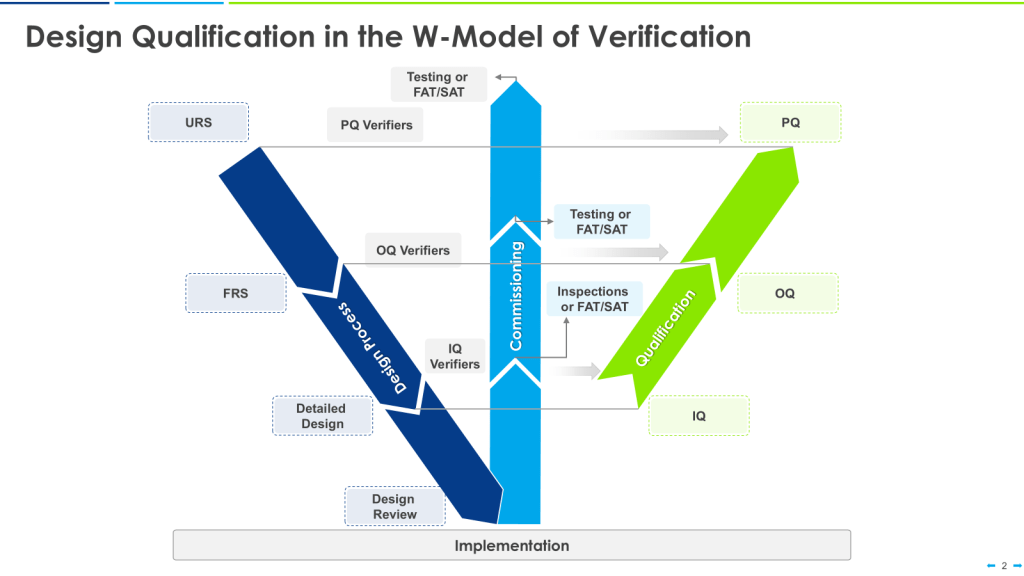
A critical step in ensuring the quality and safety of processes as part of verification is Design Review, which is sometimes expanded to Design Qualification.
Design Review is a systematic, documented examination of a proposed design to evaluate its adequacy and identify potential issues early in the development process. Here’s how to conduct an effective Design Review:
Plan Systematically: Schedule reviews at appropriate stages of development, ensuring they align with your project timeline.
Involve the Right People: Include representatives from all relevant functions and an independent reviewer not directly responsible for the design stage being evaluated.
Focus on Critical Aspects: Prioritize design elements that directly impact product quality and patient safety.
Document Thoroughly: Record all findings, including the design under review, participants, date, and any proposed actions.
Iterate as Needed: Conduct reviews iteratively as supplier design documents are published, allowing for early issue identification and correction.
Design Qualification: Verifying Suitability
Design Qualification (DQ) is the documented verification that the proposed design of facilities, equipment, or systems is suitable for its intended purpose. Here’s how to implement DQ effectively:
Develop User Requirements: Create a detailed User Requirements Specification (URS) outlining what the equipment or system is expected to do.
Create Functional Specifications: Translate user requirements into technical specifications that guide the design process.
Perform Risk Assessment: Identify potential risks associated with the design and develop mitigation strategies.
Review Design Specifications: Ensure the design meets all specified requirements, including GMP and regulatory standards.
Document and Approve: Formally document the DQ process and obtain approval from key stakeholders, including quality assurance personnel.
Integrating Design Review and DQ
To maximize the effectiveness of these processes:
Use a Risk-Based Approach: Prioritize efforts based on the level of risk to product quality and patient safety.
Leverage Subject Matter Experts: Involve SMEs from the start to contribute their expertise throughout the process.
Implement Change Management: Establish a robust system to manage design changes effectively and avoid late-stage issues.
Ensure Quality Oversight: Have Quality Assurance provide oversight to maintain compliance with current regulations and GMP requirements.
Document Comprehensively: Maintain thorough records of all reviews, qualifications, and decisions made during the process.
Implementing a systematic approach to Design Review and Design Qualification not only helps meet regulatory expectations but also contributes to operational efficiency and product excellence. As the pharmaceutical landscape evolves, staying committed to these foundational practices will remain crucial for success in this highly regulated industry.
ASTM E2500, the Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment, is intended to “satisfy international regulatory expectations in ensuring that manufacturing systems and equipment are fit for the intended use and to satisfy requirements for design, installation, operation, and performance.”
The ASTM E2500 approach is a comprehensive framework for specification setting, design, and verification of pharmaceutical and biopharmaceutical manufacturing systems and equipment. It emphasizes a risk- and science-based methodology to ensure that systems are fit for their intended use, ultimately aiming to enhance product quality and patient safety.
Despite its 17-year history, it is fair to say it is not the best-implemented standard. There are still many unrealized opportunities and some major challenges. I don’t think a single organization I’ve been in has fully aligned, and ASTM E2500 can feel aspirational.
Key Principles
Risk Management: The approach integrates risk management principles from ICH Q8, Q9, and Q10, focusing on identifying and mitigating risks to product quality and patient safety throughout the lifecycle of the manufacturing system.
Good Engineering Practices (GEP): It incorporates GEP to ensure systems are correctly designed, installed, and operated.
Flexibility and Efficiency: It strives for a more flexible and efficient organization of verification activities that can be adapted to each project’s specific context.
Regulatory agencies expect drugmakers to persuade them that we know our processes and that our facilities, equipment, systems, utilities, and procedures have been established based on concrete data and a thorough risk assessment. The ASTM E2500 standard provides a means of demonstrating that all of these factors have been validated in consideration of carefully evaluated risks.
What the Standard Calls for
Four Main Steps
Requirements: Define the system’s needs and critical aspects. Subject Matter Experts (SMEs) play a crucial role in this phase by defining needs, identifying critical aspects, and developing the verification strategy.
Specification & Design: Develop detailed specifications and design the system to meet the requirements. This step involves thorough design reviews and risk assessments to ensure the system functions as intended.
Verification: Conduct verification activities to confirm that the system meets all specified requirements. This step replaces the traditional FAT/SAT/IQ/OQ/PQ sequence with a more streamlined verification process that can be tailored to the project’s needs.
Acceptance & Release: Finalize the verification process and release the system for operational use. This step includes the final review and approval of all verification activities and documentation.
Four Cross-Functional Processes
Good Engineering Practices (GEP): Ensure all engineering activities adhere to industry standards and best practices.
Quality Risk Management: Continuously assess and manage risks to product quality and patient safety throughout the project.
Design Review: Regularly reviews the system design to ensure it meets all requirements and addresses identified risks.
Change Management: Implement a structured process for managing system changes to ensure that all modifications are appropriately evaluated and documented.
Applications and Benefits
Applicability: The ASTM E2500 approach can be applied to new and existing manufacturing systems, including laboratory, information, and medical device manufacturing systems.
Lifecycle Coverage: It applies throughout the manufacturing system’s lifecycle, from concept to retirement.
Regulatory Compliance: The approach is designed to conform with FDA, EU, and other international regulations, ensuring that systems are qualified and meet all regulatory expectations.
Efficiency and Cost Management: By focusing on critical aspects and leveraging risk management tools, the ASTM E2500 approach can streamline project execution, reduce time to market, and optimize resource utilization.
The ASTM E2500 approach provides a structured, risk-based framework for specifying, designing, and verifying pharmaceutical and biopharmaceutical manufacturing systems. It emphasizes flexibility, efficiency, and regulatory compliance, making it a valuable tool for ensuring product quality and patient safety.
What Makes it Different?
ASTM E2500
The more traditional approach
Testing Approach
It emphasizes a risk-based approach, focusing on identifying and managing risks to product quality and patient safety throughout the manufacturing system’s lifecycle. This approach allows for flexibility in organizing verification activities based on the specific context and critical aspects of the system.
Typically follows a prescriptive sequence of tests (FAT, SAT, IQ, OQ, PQ) as outlined in guidelines like EU GMP Annex 15. This method is more rigid and less adaptable to the specific needs and risks of each project.
Verification vs Qualification
The term “verification” encompasses all testing activities, which can be organized more freely and rationally to optimize efficiency. Verification activities are tailored to the project’s needs and focus on critical aspects.
Follows a structured qualification process (Installation Qualification, Operational Qualification, Performance Qualification) with predefined steps and documentation requirements.
Role of Subject Matter Experts
SMEs play a crucial role from the start of the project, contributing to the definition of needs, identification of critical aspects, system design review, and development of the verification strategy. They are involved throughout the project lifecycle.
SMEs are typically involved at specific points in the project lifecycle, primarily during the qualification phases, and may not have as continuous a role as in the ASTM E2500 approach.
Integration of Good Engineering Practices
Offers greater flexibility in organizing verification activities, allowing for a more efficient and streamlined process. This can lead to reduced time to market and optimized resource utilization.
While GEP is also important, the focus is more on the qualification steps rather than integrating GEP throughout the entire project lifecycle.
Change Management
It emphasizes early and continuous change management, starting from the supplier’s site, to avoid test duplication and ensure that changes are properly evaluated and documented.
It emphasizes early and continuous change management, starting from the supplier’s site, to avoid test duplication and ensure that changes are properly evaluated and documented.
Documentation
Documentation is focused on risk management and verification activities, ensuring compliance with international regulations (FDA, EU, ICH Q8, Q9, Q10). The approach is designed to meet regulatory expectations while allowing for flexibility in documentation.
quires extensive documentation for each qualification step, which can be more cumbersome and less adaptable to specific project needs.
Opinion
I’m watching to see what the upcoming update to Annex 15 will do to address the difficulties some see between an ATSM E2500 approach and the European regulations. I also hope we will see an update to ISPE Baseline® Guide Volume 5: Commissioning and Qualification to align an approach.
ISPE Baseline® Guide Volume 5
ATSM E2500
Design inputs Impact assessment Design Qualification Commissioning Multiple trial runs to get things right IQ, OQ, PQ, and acceptance criteria GEP Scope and QA Scope overlapped Focused on Documentation Deliverables Change Management
Design inputs Design Review Risk Mitigation Critical Control Parameters define Acceptance Criteria Verification Testing Performance Testing GEP Scope and QA Scope have a clear boundary Process, Product Quality and Patient Safety Quality by Design, Design Space, and Continuous Improvement
To be honest I don’t think ATSM E2500, ISPE Guide 5, or anything else has the balance just right. And your program ends up being a triangulation between these and the regulations. And don’t even bring in trying to align GAMP5 or USP <1058> or…or…or…
And yes, I do consider this part of my 3-year plan. I look forward to the challenges of a culture shift, increased SME involvement, formalization of GEPs (and teaching engineers how to write), effective change management, timely risk assessments, and comprehensive implementation planning.