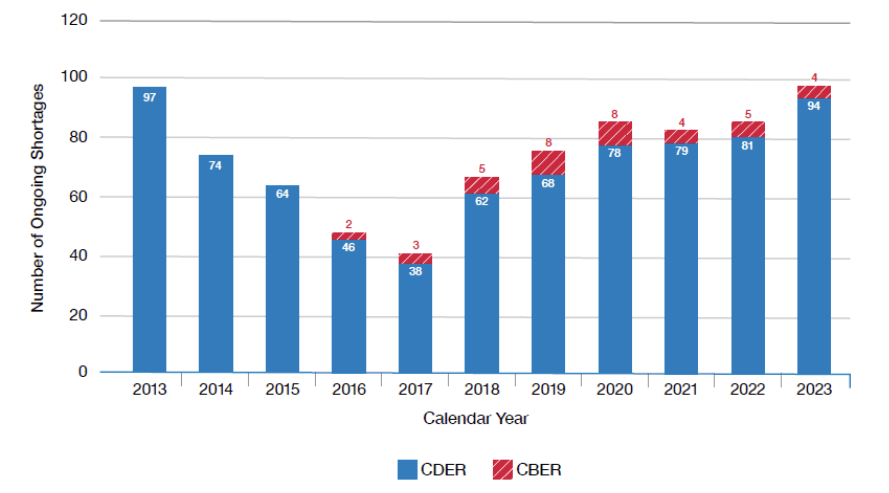
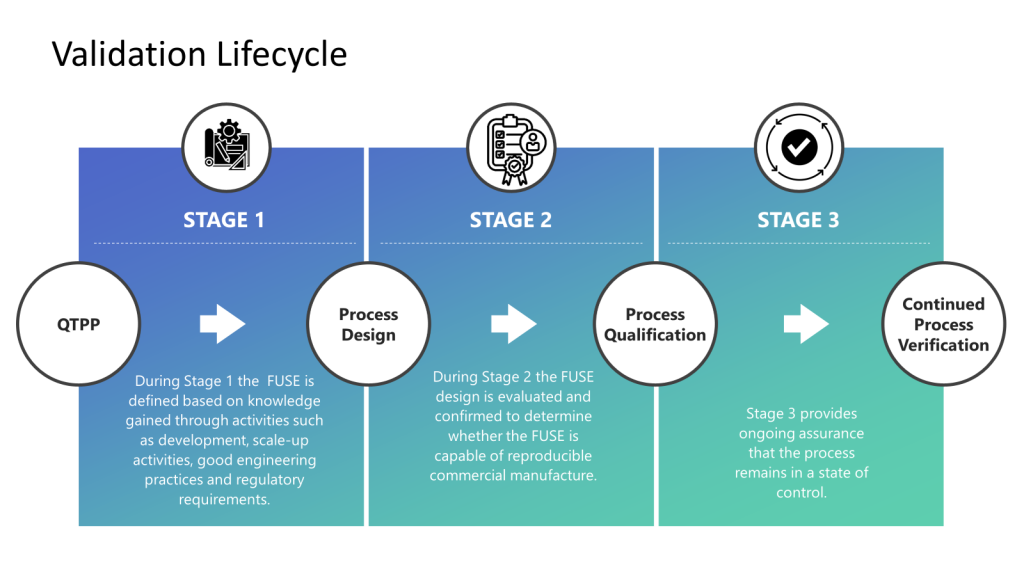
The various agency inspection manuals are critical tools for inspection readiness. I want to lay out where to find some of these manuals and then go deep into pre-approval inspections, focusing on data integrity.
European Medicines Agency
The European Medicines Agency (EMA) has established detailed procedures and work instructions for coordinating and conducting Good Clinical Practice (GCP), Good Manufacturing Practice (GMP), and pharmacovigilance inspections. Here are the key points regarding EMA’s inspection procedures:
GCP Inspection Procedures
- EMA identifies applications for GCP inspections based on risk assessment criteria and exchanges information on shared applications with the FDA.
- Inspections can be joint (conducted concurrently by EMA and FDA inspectors) or sequential (conducted separately by each agency).
- EMA notifies the applicant/marketing authorization holder (MAH) and inspects sites about upcoming inspections through the IRIS industry portal instead of formal letters.
- Applicants/MAHs must provide a signed statement accepting the inspection and granting direct access to documents and medical records.
- Requested documents should be provided directly to inspectors in electronic format after consulting the reporting inspector.
- After the inspection, EMA receives the draft inspection report, finalizes it with the inspectee’s responses, and publishes it in IRIS.
- EMA coordinates GMP inspections based on risk assessment for marketing authorization applications, variations, and routine re-inspections.
- Work instructions cover areas such as inspection announcement, fee calculation, product sampling/testing, and report circulation.
Pharmacovigilance Inspection Procedures
- EMA has specific procedures for coordinating pharmacovigilance inspections and managing non-compliance notifications from MAHs.
- Work instructions detail the inspection program creation, data entry in databases, and interactions with third-country inspectorates.
The EMA aims to harmonize inspection processes with the FDA and other regulatory bodies to streamline collaboration and information sharing while ensuring clinical trial subject protection and product quality.
FDA
The FDA Investigations Operations Manual (IOM) is the primary inspection manual used by FDA personnel when performing inspections and investigations.
The key points about the IOM are:
- It provides comprehensive instructions, procedures, and policies for FDA investigators and inspectors to follow when conducting inspections, surveys, and investigations.
- It covers inspectional activities for foods, drugs, medical devices, biologics, cosmetics, and other FDA-regulated products.
- The manual details procedures for inspections of manufacturing facilities, sampling, import operations, recalls, consumer complaints, and other compliance activities.
- It aims to ensure inspections are conducted consistently across FDA field offices and provide clear guidance to the industry on the FDA’s inspection approach.
- The IOM is updated periodically to incorporate new laws, regulations, policies, and technological changes impacting FDA’s operations.
- While not legally binding, the IOM represents the FDA’s current thinking and policies on inspections and investigations.
The FDA Investigations Operations Manual serves as the comprehensive inspection reference and procedure manual for FDA field staff carrying out the agency’s oversight and enforcement activities across all regulated product areas.
Pre-Approval Inspections
For new facilities, CPGM 7346.832, the FDA’s Compliance Program Guidance Manual for Pre-Approval Inspections (PAIs) of drug manufacturing facilities, is critical to spend time with. It outlines the objectives and procedures for FDA inspectors to evaluate a facility’s readiness for commercial manufacturing before approving a new drug application.
The key objectives of CPGM 7346.832 are:
- Assess if the facility has a quality system capable of controlling commercial manufacturing operations.
- Verify that the manufacturing processes, formulation, and analytical methods conform to the application details.
- Audit raw data integrity to authenticate the data submitted in the application.
- Evaluate the facility’s commitment to quality in pharmaceutical development (new objective added in 2022 revision).
The guidance instructs inspectors on evaluating the firm’s quality systems, process validation, data integrity, laboratory controls, change management, investigations, batch release procedures, and compliance with current Good Manufacturing Practices (cGMPs). It aims to ensure the facility can reliably produce the drug product described in the application.
Data Integrity
CPGM 7346.832 has specific requirements for data integrity audits during drug manufacturing facility pre-approval inspections (PAIs). Utilizing this document is an excellent way to evaluate your data integrity program.
The key points are:
- Objective 3 of the guidance is “Data Integrity Audit”—auditing and verifying raw data associated with the product to authenticate the data submitted in the application.
- Inspectors must audit the accuracy and completeness of data reported by the facility for the product. This involves verifying the factual integrity (data matches what was submitted) and contextual integrity (supporting data is complete).
- Inspectors should examine raw data, such as chromatograms, analyst notebooks, electronic data, etc., and compare it to the summary data in the application’s Chemistry, Manufacturing, and Controls (CMC) section.
- The data integrity audit should focus on finished product stability, dissolution, content uniformity, API impurities, etc.
- Inspectors must identify any unreported relevant data, data falsification, improper invalidation of results, or unexplained data discrepancies.
- Indications of data integrity issues include altered raw data, references to failing studies, discrepancies between samples, and missing records.
The data integrity audit aims to ensure the CMC data submitted to FDA is complete, reliable, and can be fully authenticated from the raw data at the manufacturing site. Robust data integrity is critical for the FDA to decide on the application’s approval.