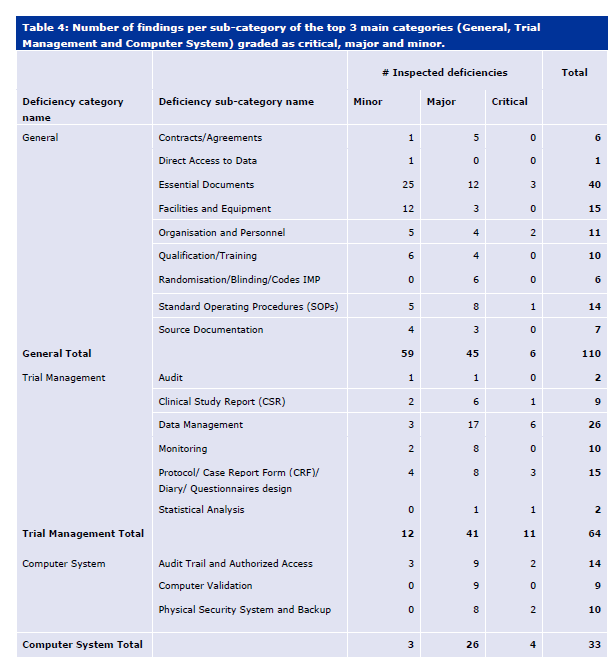
With ICH E6(r3) in draft, I think it is important to look at the current state of risk management expectations in a clinical study.
Risk management is an essential part of any clinical study, and is a critical component of the ICH E6 and E8 guidelines for Good Clinical Practice (GCP). These guidelines provide a framework for ensuring the safety and well-being of study participants, as well as the integrity and reliability of the study data. By following the principles outlined in these guidelines, researchers can help to ensure that their study results are reliable and can be used to inform clinical practice.
Through risk management we ensure the four main goals of the GCPs are obtained.

The ICH E6 guideline provides recommendations for the conduct of clinical trials, emphasizing the importance of risk management, specifying that a risk management plan should be developed and implemented for each study. The guideline also provides recommendations for the content of the risk management plan, including the identification of potential risks, the assessment of their likelihood and potential impact, and the development of strategies for managing or mitigating those risks.
Risk management is a key enabler and result of the quality management system.
The ICH E8 guideline, which focuses on the conduct of clinical trials also emphasizes the importance of risk management. It specifies that the risk management plan should include a comprehensive evaluation of the risks associated with the study interventions, as well as a plan for managing or mitigating those risks. The guideline also recommends that the risk management plan be regularly reviewed and updated as needed, to ensure that it continues to effectively address the risks facing the study.
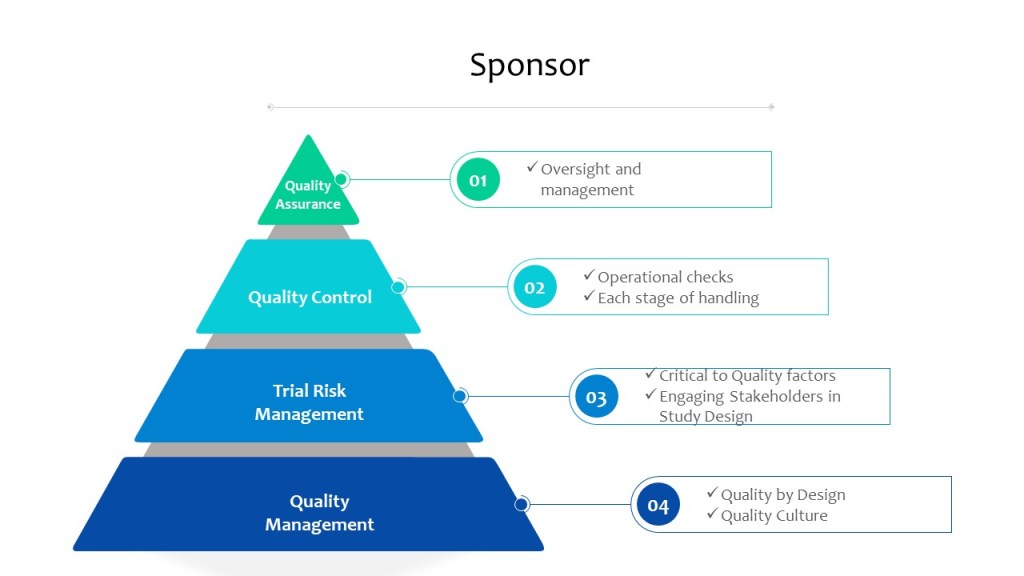
When planning a clinical study, sponsors must carefully consider the potential risks involved and take steps to minimize them. Sources of the risk assessment include performing a thorough literature review to identify any known risks associated with the study interventions, as well as conducting pre-study assessments to identify potential risks specific to the study population. E8 also state sthe importance of a wide variety of stakeholders, including the patient population.
Once the study is underway, it’s important to closely monitor for potential risks and have a plan in place for managing them.
In addition to protecting the safety of study participants, effective risk management is also essential for maintaining the integrity of the data being collected. Risks to the study data might include things like errors in data entry or missing data, which can compromise the validity of the study results. To address these risks, sponsors must have robust quality control measures in place, such as regular data audits and checks for missing or inconsistent data.
Overall, the role of risk management in a clinical study is to ensure the safety and well-being of study participants, while also protecting the integrity of the data being collected. By carefully considering and managing potential risks, researchers can help to ensure that their study results are reliable and can be used to inform clinical practice.
Risk Based Monitoring
Risk-based monitoring is a approach to monitoring the quality of a clinical study that focuses on identifying and addressing potential risks to the study. This approach involves regularly assessing the risks associated with a study and implementing strategies to manage or mitigate those risks.
In a risk-based monitoring approach, the study team typically uses a risk register to identify and assess potential risks to the study, such as the potential for errors in data collection or analysis, or the potential for adverse events in study participants. The team then develops a plan for addressing these risks, which might involve implementing additional quality control measures or training for study staff.
During the study, the team regularly monitors for potential risks and takes action to address them as needed. This might involve conducting regular audits or reviews of the study data to identify potential errors, or monitoring the health and well-being of study participants to identify and address any adverse events.
Overall, the goal of risk-based monitoring is to ensure the quality and integrity of a clinical study by proactively identifying and addressing potential risks. By using a risk-based approach, the study team can help to ensure that the study results are reliable and can be used to inform clinical practice.
Risk Register
A risk register is a document that is used to identify, assess, and track potential risks in a clinical study. It typically includes a list of identified risks, along with information about their likelihood and potential impact, as well as the actions that are being taken to manage or mitigate the risks.
In a clinical study, a risk register might include risks such as the potential for errors in data collection or analysis, the potential for adverse events in study participants, or the potential for the study to be impacted by external factors, such as changes in regulatory requirements.
The purpose of a risk register in a clinical study is to help the study team identify and prioritize potential risks, and to develop strategies for addressing them. By having a clear and comprehensive overview of the risks that a study is facing, the team can take proactive steps to manage or mitigate those risks, and can monitor their progress over time.
Overall, a risk register is an essential tool for managing risks in a clinical study. By providing a clear and comprehensive overview of potential risks, it helps the study team identify and address risks in a proactive and effective way.
- Identifying potential risks: The first step in implementing a clinical risk management program is to identify potential risks to the study, such as the potential for errors in data collection or analysis, or the potential for adverse events in study participants. This might involve reviewing the study protocol and data collection tools, consulting with the study team and other stakeholders, and conducting a thorough assessment of the study environment.
- Assessing risks: Once potential risks have been identified, the next step is to assess their likelihood and potential impact. This will help to prioritize the risks and determine the appropriate level of response. For example, a risk with a high likelihood and a high potential impact might require more immediate action, while a risk with a low likelihood and a low potential impact might not require as much attention.
- Developing strategies for managing risks: Based on the assessment of risks, the next step is to develop strategies for managing or mitigating those risks. This might involve implementing additional quality control measures, providing training to study staff, or conducting regular audits or reviews of the study data. The goal is to develop a comprehensive and effective plan for addressing the identified risks.
- Monitoring for potential risks: Once the risk management plan is in place, it’s important to regularly monitor for potential risks and take action to address them as needed. This might involve conducting regular audits or reviews of the study data, or monitoring the health and well-being of study participants. By proactively monitoring for potential risks, the study team can help to ensure the safety and well-being of study participants, as well as the integrity and reliability of the study data.
- Follow-up and corrective action: If potential risks are identified during the study, it’s important to take prompt action to address them. This might involve implementing corrective action plans, such as retraining study staff or revising the study protocol. It’s also important to track the progress of these plans and ensure that they are effective in addressing the identified risks. By taking timely and effective action to address potential risks, the study team can help to ensure the safety and well-being of study participants, as well as the integrity and reliability of the study data.
Risk Management in the Clinical Study Process
To summarize, each clinical study should:
- Identify Risks
- Before the study begins, the sponsor should perform a thorough review of the study protocol, data collection tools, and other study-related documents to identify potential risks to the study.
- The cross-functional study team, CROs and other relevant stakeholders, such as the sponsor and regulatory authorities, to identify additional potential risks.
- All identified risks should be documented in the study’s risk register.
2. Assess Risks
- For each identified risk, assess its likelihood and potential impact on the study.
- The risks should be prioritized based on their likelihood and potential impact, with a focus on the highest-priority risks.
3. Manage Risks
- For each identified risk, the sponsor should develop a plan for managing or mitigating the risk. This plan should be documented in the study’s risk register.
- The plan for managing or mitigating each risk should include specific actions to be taken, as well as the individuals or groups responsible for implementing those actions.
4. Monitor Risks
- Regularly monitor key risk indicators and the study for success of the study risk plan and to identify new potential risks and take action to address them as needed. This might involve conducting regular audits or reviews of the study data, or monitoring the health and well-being of study participants.
- Any significant risks that arise during the study should be reported to the sponsor and relevant regulatory authorities.