The FDA’s reorganization has been unveiled and will be implemented on October 1st. As a total wonk, this is exciting.
There are two major changes:
Forming a Human Food Program (HFP) to consolidate a preventive approach will not have much impact on me professionally, but I’m hoping that as a consumer, we see significant dividends from this refocus.
ORA is being renamed the Office of Inspections and Investigations (OII) and will focus on inspections, investigations, and imports as its core mission. If nothing else, this will make explaining the structure of the FDA a heck of a lot easier.
Everything else seems to be mostly a lot of shuffling of the deck chairs that will have little impact.
Assure data integrity in the context of GMP. This would be in parallel with similar consideration of Annex 11 (Computerised Systems).
Q1 2026
An update is needed to align with current thinking. Data Integrity has advanced significantly in the last five years, and Chapter 4 could benefit from alignment with the PIC/S guidance.
GMP Guide: Annex 11 (Computerised Systems)
Assure data integrity in the context of GMP. This would be in parallel with similar consideration of Chapter 4 (Documentation).
Q1 2026
A necessary update. Will be curious to see how it aligns with the FDA’s CSA approach (which isn’t really all that new).
We pretty much know what will be in it from the concept paper. At least it will solidify this requirement for cloud systems “Regulated users should 26 have access to the complete documentation for validation and safe operation of a system and be able to present this during regulatory inspections, e.g. with the help of the service provider.”
Guidelines on GMP specific to ATMPS
Review the Guidelines in collaboration with CAT and the European Commission following the publication of a new regulation on standards of quality and safety for substances of human origin intended for human application and need to update legal references and definitions. Review the Guidelines in the light of new Annex 1 Manufacture of Sterile Medicinal Products and consider whether any updates are necessary.
Q4 2026
This is a fast area of change, and this update is called for.
Aligning to Annex 1 is overdue.
GMP Guide: Annex 3 Manufacture of Radiopharmaceuticals
A review and update of the Annex to reflect current state of the art.
Q4 2026
I’ve never worked in radiopharmaceuticals. Maybe someday.
GMP Guide: Annex 15 Qualification and Validation
In the context of new technology in facilities, products and processes and following up on LLE recommendations, and extend the scope to APIs.
Q4 2025
LLE is the EMA’s lessons learnt report (LLE) on Nitrosamines.
I’d love to see significant changes to finally align with ATSM E2500 and other recent challenges in validation.
GMP Guide: Annex 16 Certification by a Qualified Person and Batch Release
Following up on LLE recommendations.
Q4 2025
I’m not a massive fan of QPs as structured. Not expecting that to change.
GMP and Marketing Authorisation Holders
To revise the paper in line with recommendations from the Nitrosamines LLE, to strengthen guidance for MAHs in terms of having adequate quality agreement with manufactures.
Q4 2025
Anything to strengthen quality agreements is probably a good thing.
Anytime we see a major chapter update in the Eudralex Volume 4 is an exciting year, and the next few promise to be big. Maybe not Annex 1 big, but maybe the EMA and PIC/S will surprise us.
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Chapter 3: Premises and Equipment, (2014)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Chapter 5: Production, (2014)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Part II: Basic Requirements for Active Substances used as Starting Materials, (2014)
European Union, Guidelines of 19 March 2015 on the formalized risk assessment for ascertaining the appropriate good manufacturing practice for excipients of medicinal products for human use, Official Journal of the European Union, (2015/C 95/02), (2015)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Annex 2: Manufacture of Biological active substances and Medicinal Products for Human Use, (2018)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Annex 3 Manufacture of Radiopharmaceuticals, (2008)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Annex 14 Manufacture of Medicinal Products Derived from Human Blood or Plasma, (2011)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products, (2017)
European Union, Guidelines of 5 November 2013 on Good Distribution Practice of medicinal products for human use, Official Journal of the European Union, (2013/C 343/01), (2013),
European Union, Guidelines of 19 March 2015 on principles of Good Distribution Practice of active substances for medicinal products for human use, Official Journal of the European Union, (2015/C 95/01), (2015)
EMA Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities (20 November 2014)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, subpart C = Building and Facilities, sec. 211.42 Design and construction features (b), (c)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart F – Production and Process Controls, sec. 211.113 Control of microbial contamination (a), (b)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart B – Organization and Personnel, sec.211.28 Personnel responsibilities (a)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart E – Control ofComponents and Drug Product Containers and Closures, sec. 211.80 General requirements. (b)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart E – Control of Components and Drug Product Containers and Closures, sec. 211.84 Testing and approval or rejection of components, drug product containers, and closures (d)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart D – Equipment, sec.211.67 Equipment cleaning and maintenance (a)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart C – Buildings and Facilities, sec. 211.56 Sanitation (c)
U.S. Food & Drug Administration, Guidance for Industry Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice, (2004)
U.S. Food & Drug Administration, Guidance for Industry – Good Manufacturing Practice Considerations for Responding to COVID-19 Infection in Employees in Drug and Biological Products Manufacturing, (2020)
U.S. Food & Drug Administration, Guidance for Industry – Guidance for Industry Non-Penicillin Beta-Lactam Drugs: A CGMP Framework for Preventing Cross Contamination, (2013)
U.S. Food & Drug Administrationn, Guidance for Industry Current Good Manufacturing Practice—Guidance for Human Drug Compounding Outsourcing Facilities Under Section 503B of the FD&C Act, Draft Guidance. https://www.fda.gov/media/88905/download (accessed Mar 6, 2022)
Pharmaceutical Inspection Co-operation Scheme gmp guide, 2nd targeted consultation document on revision of annex 1
Pharmaceutical Inspection Co-operation Schemepharmaceutical inspection co-operation scheme gmp guide, ps inf 25 2019 (rev. 1) draft, manufacture of advanced therapy medicinal products for human use
Pharmaceutical Inspection Co-operation Scheme gmp guide, ps inf 26 2019 (rev. 1) draft, manufacture of biological medicinal substances and products for human use
Pharmaceutical Inspection Co-operation Scheme gmp guide, pe 009-15 (part i), guide to good manufacturing practice for medicinal products part i
Pharmaceutical Inspection Co-operation Scheme gmp guide, pe 009-15 (part ii), guide to good manufacturing practice for medicinal products part ii
Pharmaceutical Inspection Co-operation Scheme gmp guide, pe 009-15 (annexes), guide to good manufacturing practice for medicinal products annexes
World Health Organisation, good manufacturing practices for pharmaceutical products: main principles, annex 2, who technical report series 986, 2014,
World Health Organisation, who good manufacturing practices for active pharmaceutical ingredients (bulk drug substances), annex 2, who technical report series 957, 2010
World Health Organisation, points to consider for manufacturers and inspectors: environmental aspects of manufacturing for the prevention of antimicrobial resistance annex 6, who technical report series 1025, 2020
World Health Organisation, WHO good manufacturing practices for sterile pharmaceutical products, annex 6, who technical report series 961, 2011
World Health Organisation, WHO good manufacturing practices for biological products, annex 3, who technical report series 996, 2016
World Health Organisation, WHO good manufacturing practices for the manufacture of investigational pharmaceutical products for clinical trials in humans, annex 7, who technical report series 863, 1996
World Health Organisation, WHO good manufacturing practices for radiopharmaceutical products annex 2, who technical report series 1025, 2020
World Health Organisation, WHO GMP for Pharmaceutical Products containing Hazardous Substances, TRS 957, Annex-3 (2010)
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human use, Quality Risk Management, Q8 (R2), Pharmaceutical Development, August 2009. https://database.ich.org/sites/default/files/Q8%28R2%29%20Guideline.pdf (Accessed Mar 06, 2022)
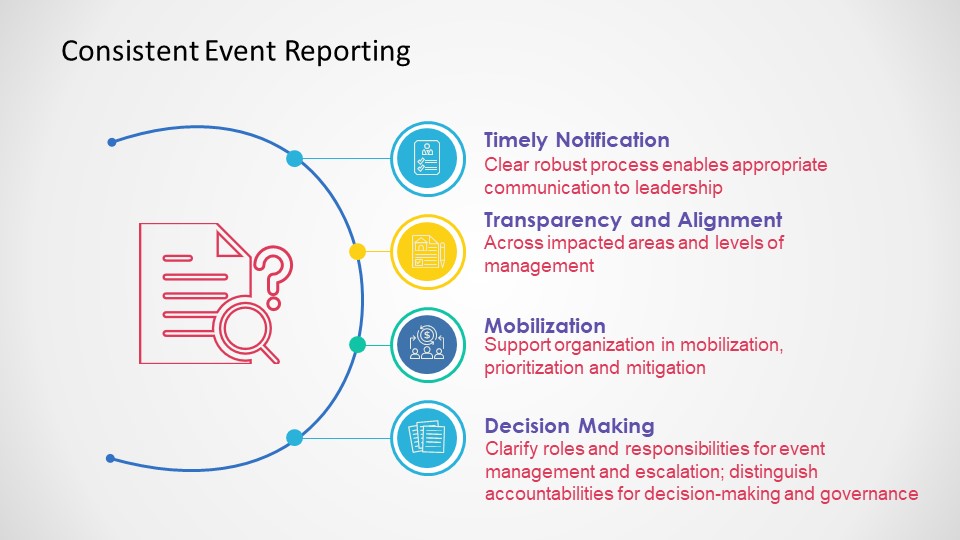
Event management systems need to have an escalation mechanism to ensure critical events are quickly elevated to a senior level to ensure organization-wide timely reactions.
Consistent Event Reporting
There are many reasons for a fast escalation.
Events that trigger reporting to Regulatory Agencies (e.g. Serious Breach, Urgent Safety Measures (UK), Field Alerts, Biological Product Deviation, Medical Device Report)
Events that require immediate action to prevent additional harm from across the organization
Events that require marshalling resources from large parts of the organization
•Reference GxP area for Impact
resulting from/linked to system error/failure
•Product Quality/ CMC events in
accordance with MRB criteria (or other events of similar scope of impact)
•Impact to study integrity
•Impact to subject’s safety, rights or
welfare
•Gaps in reporting/ collection of
potential AEs
•Impact to study integrity
•Impact to study integrity
•System design, testing, deployment,
upgrade, etc. event impacting GxP data integrity or regulatory compliance
•Recurring event with broad scope of
impact
•Recurring event with broad scope of
impact
•Recurring event with broad scope of
impact
•Recurring event with broad scope of
impact
•Recurring event with broad scope of
impact
•Recurring event with broad scope of
impact
•Impact to program milestones & corporate
goals
•Impact to program milestones & corporate
goals
•Impact to program milestones & corporate
goals
•Impact to program milestones & corporate
goals
•Impact to program milestones & corporate
goals
•Potential Falsified or Counterfeit
Product
•Potential Fraud or Misconduct
•Potential Fraud or Misconduct
•Credible Risk of Product Shortage
•Quality event with patient safety
risk/gap
•GxP Data Breach
•Potential Product Recall
•Significant Quality Event Notified to
Regulatory Authority
•System error or failure with
significant GxP compliance impact
·Potential Critical Finding Resulting from
Regulatory Authority Inspection or Audit by External Body/Third Party
·Quality Event/Observation Classified
as Critical (Event or Internal Audit) Notification from Regulatory Authority
or other External Authority of Findings of Significant/Critical Quality
Deficiency (inspection or other than through inspection)
oe.g.; Refusal to File, Notification
of Inadequate Response to Inspection Findings (e.g.; Other Action Indicated
(FDA classification), Warning Letter
You can drill down to a lower, more practical level, like this
Escalation Criteria
Examples of Quality Events for Escalation
Potential to adversely affect
quality, safety, efficacy, performance or compliance of product (commercial
or clinical)
•Contamination (product, raw material,
equipment, micro; environmental)
•Product defect/deviation from process
parameters or specification (on file with agencies)
•Significant GMP deviations
•Incorrect/deficient labeling
•Product complaints (significant PC,
trends in PCs)
•OOS/OOT (e.g., stability)
Product counterfeiting, tampering, theft
•Product counterfeiting, tampering, theft reportable to Health
Authority (HA)
•Lost/stolen IMP
•Fraud or misconduct associated with counterfeiting, tampering,
theft
•Potential to impact product supply (e.g., removal, correction,
recall)
Product shortage likely to
disrupt patient care and/or reportable to HA
•Disruption of product supply due to
product quality events, natural disasters (business continuity disruption),
OOS impact, capacity constraints
Potential to cause patient harm associated with a product
quality event
•Urgent Safety Measure, Serious Breach, Significant Product
Compliant, Safety Signal that are determined associated with a product
quality event
Significant GMP
non-compliance/event
•Non-compliance or non-conformance
event with potential to impact product performance meeting specification,
safety efficacy or regulatory requirements
Regulatory Compliance Event
•Significant (critical, repeat) regulatory inspection findings,
lack of commitment adherence
•Notification of directed/for cause inspection
•Notification of HA correspondence indicating potential
regulatory action
An updated and expanded version of this is found here.
Primary Investigator, Study Director, Qualified Person, Responsible Person – the pharmaceutical regulations are rife with a series of positions that are charged with achieving compliance and quality results. I tend to think of them as a giant Achilles heel created by the regulations.
The concept of an individual having all the accountability is nowhere near universal, for example, the term Quality Unit is a nice inclusive we – though I do have some quibbles on how it can end up placing the quality unit within the organization.
This is an application of the great man fallacy – the idea that one person by the brunt of education, experience, and stunning good looks can ensure product safety, efficacy and quality, and all the other aspects of patient and data integrity of trials.
That is, frankly, poppycock.
People only perform successfully when they are in a well-built system. Process drives success and leverages the right people at the right time making the right decisions with the right information. No one person can do that, and frankly thinking someone can is setting them up for failure. Which we see, a lot in the regulatory space.
Sure, the requirement exists, we need to meet it failing the agencies waking up and realizing the regulations are setting us up for failure. But we don’t need to buy into it. We build our processes to leverage the team, to democratize decisions, and to drive for reliable results.
Let’s leave the great man theory in the dustbins where it belongs.