A familiar scene exists across every pharmaceutical manufacturing site I’ve ever seen, lot disposition cycle times are a struggle. While management instinctively pushes for “optimization everywhere,” the quality department remains overwhelmed and becomes the weakest link in an otherwise robust chain. This scenario illustrates perfectly why understanding and applying the Theory of Constraints (TOC) is essential for quality excellence in complex systems.
The Fundamentals of Theory of Constraints
The Theory of Constraints, developed by management guru Eliyahu M. Goldratt in his groundbreaking 1984 book The Goal, fundamentally changed how we view process improvement. Unlike approaches that attempt to optimize all parts of a system simultaneously, TOC recognizes a profound truth: in any system, there is always at least one constraint-a bottleneck-that limits overall performance. This constraint determines the maximum throughput of the entire system, regardless of how efficient other components might be.
TOC defines a constraint as “anything that prevents the system from achieving its goal,” which in business typically translates to generating profit but can also be viewed as getting product to the patient. By focusing improvement efforts specifically on these constraints rather than dispersing resources across the system, organizations can achieve more significant results with less effort. This laser-focused approach makes TOC not just another quality tool but a foundational framework that bridges system thinking with practical quality management.
The Power of the Weakest Link Paradigm
Systems thinking teaches us that organizations are networks of interdependent processes in which the performance of the whole exceeds the sum of its parts. TOC enhances this perspective by providing a clear mechanism for prioritization. As Goldratt famously observed, “a chain is only as strong as its weakest link.” This metaphor eloquently captures the essence of constraint management-no matter how much you strengthen other links, the chain’s overall strength remains limited by its weakest component.
This perspective fundamentally challenges the traditional approach of seeking balanced capacity across all processes.
The Five Focusing Steps: A Systematic Approach to Constraint Management
The heart of TOC’s practical application lies in the Five Focusing Steps-a powerful cyclic methodology that systematically addresses constraints:
Identify the system’s constraint(s): Determine what limits the system’s performance.
Decide how to exploit the constraint: Maximize the efficiency of the constraint without major investments.
Subordinate everything else to the above decision: Align all other processes to support the constraint’s optimal performance.
Elevate the system’s constraint: If necessary, make larger investments to increase the constraint’s capacity.
Warning! If in the previous steps a constraint has been broken, go back to step 1, but don’t allow inertia to create a new constraint: Once a constraint is resolved, the improvement cycle begins again with the new limiting factor.
This approach aligns perfectly with the system thinking principles outlined in “Principles behind a good system,” which highlight balance, coordination, and sustainability as critical elements of well-designed systems. The systematic nature of TOC provides a clear roadmap for addressing complex system challenges without becoming overwhelmed by their complexity.
TOC, Lean, and Six Sigma: A Powerful Triad
While TOC focuses on constraints, Lean targets waste elimination, and Six Sigma concentrates on reducing variation. Rather than competing methodologies, these approaches complement each other in what some practitioners call “TLSS” (TOC, Lean, Six Sigma).
The synergy becomes evident when we consider their respective objectives:
Methodology
Primary Focus
Key Metric
Philosophy
TOC
Bottlenecks
Throughput
“Find the constraint. Fix it. Repeat.”
Lean
Waste
Value Flow
“If it doesn’t add value, it’s waste.”
Six Sigma
Variation
Quality
“Reduce variation to meet customer expectations.”
TOC says ‘What’s broken?’ Lean says ‘Here’s how to fix it right.'” This complementary relationship makes TOC particularly valuable as a prioritization mechanism for quality improvement initiatives-pointing precisely where Lean and Six Sigma tools should be applied for maximum impact.
Constraints, Waste, and Variation: An Interconnected Trilogy
Constraints in a system often become amplifiers of waste and variation. When a process operates at capacity, minor variations become magnified, and waste becomes more impactful. Consider a quality testing laboratory operating at its constraint-even small variations in testing time or minor errors requiring rework can cascade into significant delays, exacerbating waste throughout the system.
This interconnection helps explain why constraint management must be integrated with waste reduction and variation control. The goal is not just to fix immediate issues but to prevent recurrence and drive continuous improvement. TOC provides the critical prioritization framework to ensure these improvement efforts target the most impactful areas.
Throughput as a Quality Metric: Beyond Efficiency to Effectiveness
TOC introduces a clear set of metrics that differ from traditional accounting measures: throughput (the rate at which the system generates money through sales), inventory (all the money invested in things intended to be sold), and operating expense (all money spent turning inventory into throughput).
This focus on throughput as the primary metric represents a significant shift in quality thinking. Rather than optimizing local metrics or cost-cutting, TOC emphasizes increasing the flow of value through the system-aligning perfectly with the concept of operational stability as “the state where manufacturing and quality processes exhibit consistent, predictable performance over time with minimal unexpected variations”. This emphasis on flow over efficiency helps organizations maintain focus on outcomes rather than activities.
TOC in Quality Maturity: A Path to Excellence
From Constraint Neglect to Strategic Constraint Management
Quality maturity models provide a roadmap for organizational improvement, and TOC can be mapped to these models to illustrate progression in constraint management capability:
Level 1: Initial (Constraint Neglect)
At this level, constraints are neither identified nor managed systematically. The organization experiences frequent firefighting and may attempt to “optimize” all processes simultaneously, resulting in scattered efforts and minimal system improvement. Quality issues are addressed reactively, much like the early stages of validation programs described as “ad hoc and lacking standardization”.
Level 2: Managed (Constraint Awareness)
Organizations at this level recognize the existence of constraints but address them in silos. There’s increased awareness of bottlenecks, but responses remain tactical rather than strategic. This parallels the “Managed” validation maturity level where “basic processes are established but may not fully align with guidelines”. Constraints are managed as isolated problems rather than system limitations.
Level 3: Standardized (Constraint Management)
At this level, constraint identification and management become standardized across the organization. The Five Focusing Steps are consistently applied, and there’s alignment between constraint management and other quality initiatives. This mirrors the “Standardized” level in validation maturity where “processes are well-defined and consistently implemented”.
Organizations at this level not only manage current constraints but predict future ones through data analysis. Constraint metrics are established and regularly monitored, similar to the “Predictable” validation maturity level where “KPIs for validation activities are established and regularly monitored”.
At the highest maturity level, constraint management becomes embedded in strategic planning. The organization continuously innovates its approach to constraints and may actively design systems to control where constraints appear. This aligns with the “Optimizing” validation maturity level characterized by “continuous improvement and innovation.”
This maturity progression illustrates how TOC implementation evolves from reactive problem-solving to strategic system design, paralleling broader quality maturity development.
Actionable Insights: Implementing TOC in Your Quality System
Step 1: Map Your Value Stream to Identify Potential Constraints
Process mapping is a fundamental first step in constraint identification. As noted in “Process Mapping as a Scaling Solution,” a process flow diagram is a visual representation of a process’s steps, showing the sequence of activities from start to finish. This visualization helps identify where materials, information, or approvals might be bottlenecked.
When mapping your value stream, pay particular attention to:
Where work accumulates or waits
Processes with high utilization rates
Steps requiring specialized resources or expertise
Points where batching occurs
Areas with high rework rates
Step 2: Analyze System Performance to Confirm the Constraint
Once potential constraints are identified, analyze performance data to confirm where the true system constraint lies. Remember, as TOC teaches, “organizations have very few true constraints.” Look for:
Processes that are consistently running at capacity.
Steps that dictate the pace of the entire system
Areas where expediting frequently occurs
Processes that, when improved, directly improve overall system performance
Step 3: Apply the Five Focusing Steps
With the constraint identified, systematically apply the Five Focusing Steps:
Identify: Document exactly what limits the constraint’s performance.
Exploit: Before investing in expansion, ensure the constraint operates at maximum efficiency. For example, in a quality testing lab constraint, this might mean eliminating administrative delays, optimizing scheduling, and ensuring the constraint never waits for inputs.
Subordinate: Adjust all other processes to support the constraint. This might include changing batch sizes, scheduling, or staffing patterns in non-constraint areas to ensure the constraint never starves or becomes blocked.
Elevate: Only after fully exploiting the constraint should you invest in expanding its capacity through additional resources, technology, or process redesign.
Repeat: Once the constraint is no longer limiting system performance, a new constraint will emerge. Return to step one to identify this new constraint.
Step 4: Integrate TOC with Your CAPA System
TOC provides an excellent framework for prioritizing corrective and preventive actions. As noted in discussions of CAPA systems, “one reason to invest in the CAPA program is that you will see fewer deviations over time as you fix issues.” By focusing CAPA efforts on constraints, you maximize the system-wide impact of improvements.
This approach ensures your quality improvement efforts focus on areas that will most significantly improve overall system performance.
Conclusion: TOC as a Quality Mindset
The Theory of Constraints offers more than just a methodology for improvement-it represents a fundamental shift in how we think about system performance and quality management. By recognizing that systems are inherently limited by constraints and systematically addressing these limitations, organizations can achieve breakthrough improvements with focused effort.
As quality systems mature, the integration of TOC principles becomes increasingly important. From reactive problem-solving to proactive constraint management and ultimately to strategic constraint design, TOC provides a path to quality excellence that complements and enhances other methodologies.
The journey to quality maturity requires system thinking, disciplined focus, and continuous improvement-all principles embodied in the Theory of Constraints. By adopting TOC not just as a tool but as a mindset, quality professionals can navigate the complexity of modern systems with clarity and purpose, ensuring resources are directed where they will have the greatest impact.
I invite you to explore more about integrating TOC with quality systems in related posts on system thinking principles, operational stability, and maturity models. The constraint may be your system’s limitation-but identifying it is your greatest opportunity for breakthrough improvement.
The pharmaceutical industry has long operated under a fundamental epistemological fallacy that undermines our ability to truly understand the effectiveness of our quality systems. We celebrate zero deviations, zero recalls, zero adverse events, and zero regulatory observations as evidence that our systems are working. But a fundamental fact we tend to ignore is that we are confusing the absence of evidence with evidence of absence—a logical error that not only fails to prove effectiveness but actively impedes our ability to build more robust, science-based quality systems.
This challenge strikes at the heart of how we approach quality risk management. When our primary evidence of “success” is that nothing bad happened, we create unfalsifiable systems that can never truly be proven wrong.
The Philosophical Foundation: Falsifiability in Quality Risk Management
Karl Popper’s theory of falsification fundamentally challenges how we think about scientific validity. For Popper, the distinguishing characteristic of genuine scientific theories is not that they can be proven true, but that they can be proven false. A theory that cannot conceivably be refuted by any possible observation is not scientific—it’s metaphysical speculation.
Applied to quality risk management, this creates an uncomfortable truth: most of our current approaches to demonstrating system effectiveness are fundamentally unscientific. When we design quality systems around preventing negative outcomes and then use the absence of those outcomes as evidence of effectiveness, we create what Popper would call unfalsifiable propositions. No possible observation could ever prove our system ineffective as long as we frame effectiveness in terms of what didn’t happen.
Consider the typical pharmaceutical quality narrative: “Our manufacturing process is validated because we haven’t had any quality failures in twelve months.” This statement is unfalsifiable because it can always accommodate new information. If a failure occurs next month, we simply adjust our understanding of the system’s reliability without questioning the fundamental assumption that absence of failure equals validation. We might implement corrective actions, but we rarely question whether our original validation approach was capable of detecting the problems that eventually manifested.
Most of our current risk models are either highly predictive but untestable (making them useful for operational decisions but scientifically questionable) or neither predictive nor testable (making them primarily compliance exercises). The goal should be to move toward models are both scientifically rigorous and practically useful.
This philosophical foundation has practical implications for how we design and evaluate quality risk management systems. Instead of asking “How can we prevent bad things from happening?” we should be asking “How can we design systems that will fail in predictable ways when our underlying assumptions are wrong?” The first question leads to unfalsifiable defensive strategies; the second leads to falsifiable, scientifically valid approaches to quality assurance.
Why “Nothing Bad Happened” Isn’t Evidence of Effectiveness
The fundamental problem with using negative evidence to prove positive claims extends far beyond philosophical niceties, it creates systemic blindness that prevents us from understanding what actually drives quality outcomes. When we frame effectiveness in terms of absence, we lose the ability to distinguish between systems that work for the right reasons and systems that appear to work due to luck, external factors, or measurement limitations.
Scenario
Null Hypothesis
What Rejection Proves
What Non-Rejection Proves
Popperian Assessment
Traditional Efficacy Testing
No difference between treatment and control
Treatment is effective
Cannot prove effectiveness
Falsifiable and useful
Traditional Safety Testing
No increased risk
Treatment increases risk
Cannot prove safety
Unfalsifiable for safety
Absence of Events (Current)
No safety signal detected
Cannot prove anything
Cannot prove safety
Unfalsifiable
Non-inferiority Approach
Excess risk > acceptable margin
Treatment is acceptably safe
Cannot prove safety
Partially falsifiable
Falsification-Based Safety
Safety controls are inadequate
Current safety measures fail
Safety controls are adequate
Falsifiable and actionable
The table above demonstrates how traditional safety and effectiveness assessments fall into unfalsifiable categories. Traditional safety testing, for example, attempts to prove that something doesn’t increase risk, but this can never be definitively demonstrated—we can only fail to detect increased risk within the limitations of our study design. This creates a false confidence that may not be justified by the actual evidence.
The Sampling Illusion: When we observe zero deviations in a batch of 1000 units, we often conclude that our process is in control. But this conclusion conflates statistical power with actual system performance. With typical sampling strategies, we might have only 10% power to detect a 1% defect rate. The “zero observations” reflect our measurement limitations, not process capability.
The Survivorship Bias: Systems that appear effective may be surviving not because they’re well-designed, but because they haven’t yet encountered the conditions that would reveal their weaknesses. Our quality systems are often validated under ideal conditions and then extrapolated to real-world operations where different failure modes may dominate.
The Attribution Problem: When nothing bad happens, we attribute success to our quality systems without considering alternative explanations. Market forces, supplier improvements, regulatory changes, or simple random variation might be the actual drivers of observed outcomes.
Observable Outcome
Traditional Interpretation
Popperian Critique
What We Actually Know
Testable Alternative
Zero adverse events in 1000 patients
“The drug is safe”
Absence of evidence does not equal Evidence of absence
No events detected in this sample
Test limits of safety margin
Zero manufacturing deviations in 12 months
“The process is in control”
No failures observed does not equal a Failure-proof system
No deviations detected with current methods
Challenge process with stress conditions
Zero regulatory observations
“The system is compliant”
No findings does not equal No problems exist
No issues found during inspection
Audit against specific failure modes
Zero product recalls
“Quality is assured”
No recalls does not equal No quality issues
No quality failures reached market
Test recall procedures and detection
Zero patient complaints
“Customer satisfaction achieved”
No complaints does not equal No problems
No complaints received through channels
Actively solicit feedback mechanisms
This table illustrates how traditional interpretations of “positive” outcomes (nothing bad happened) fail to provide actionable knowledge. The Popperian critique reveals that these observations tell us far less than we typically assume, and the testable alternatives provide pathways toward more rigorous evaluation of system effectiveness.
The pharmaceutical industry’s reliance on these unfalsifiable approaches creates several downstream problems. First, it prevents genuine learning and improvement because we can’t distinguish effective interventions from ineffective ones. Second, it encourages defensive mindsets that prioritize risk avoidance over value creation. Third, it undermines our ability to make resource allocation decisions based on actual evidence of what works.
The Model Usefulness Problem: When Predictions Don’t Match Reality
George Box’s famous aphorism that “all models are wrong, but some are useful” provides a pragmatic framework for this challenge, but it doesn’t resolve the deeper question of how to determine when a model has crossed from “useful” to “misleading.” Popper’s falsifiability criterion offers one approach: useful models should make specific, testable predictions that could potentially be proven wrong by future observations.
The challenge in pharmaceutical quality management is that our models often serve multiple purposes that may be in tension with each other. Models used for regulatory submission need to demonstrate conservative estimates of risk to ensure patient safety. Models used for operational decision-making need to provide actionable insights for process optimization. Models used for resource allocation need to enable comparison of risks across different areas of the business.
When the same model serves all these purposes, it often fails to serve any of them well. Regulatory models become so conservative that they provide little guidance for actual operations. Operational models become so complex that they’re difficult to validate or falsify. Resource allocation models become so simplified that they obscure important differences in risk characteristics.
The solution isn’t to abandon modeling, but to be more explicit about the purpose each model serves and the criteria by which its usefulness should be judged. For regulatory purposes, conservative models that err on the side of safety may be appropriate even if they systematically overestimate risks. For operational decision-making, models should be judged primarily on their ability to correctly rank-order interventions by their impact on relevant outcomes. For scientific understanding, models should be designed to make falsifiable predictions that can be tested through controlled experiments or systematic observation.
Consider the example of cleaning validation, where we use models to predict the probability of cross-contamination between manufacturing campaigns. Traditional approaches focus on demonstrating that residual contamination levels are below acceptance criteria—essentially proving a negative. But this approach tells us nothing about the relative importance of different cleaning parameters, the margin of safety in our current procedures, or the conditions under which our cleaning might fail.
A more falsifiable approach would make specific predictions about how changes in cleaning parameters affect contamination levels. We might hypothesize that doubling the rinse time reduces contamination by 50%, or that certain product sequences create systematically higher contamination risks. These hypotheses can be tested and potentially falsified, providing genuine learning about the underlying system behavior.
From Defensive to Testable Risk Management
The evolution from defensive to testable risk management represents a fundamental shift in how we conceptualize quality systems. Traditional defensive approaches ask, “How can we prevent failures?” Testable approaches ask, “How can we design systems that fail predictably when our assumptions are wrong?” This shift moves us from unfalsifiable defensive strategies toward scientifically rigorous quality management.
This transition aligns with the broader evolution in risk thinking documented in ICH Q9(R1) and ISO 31000, which recognize risk as “the effect of uncertainty on objectives” where that effect can be positive, negative, or both. By expanding our definition of risk to include opportunities as well as threats, we create space for falsifiable hypotheses about system performance.
The integration of opportunity-based thinking with Popperian falsifiability creates powerful synergies. When we hypothesize that a particular quality intervention will not only reduce defects but also improve efficiency, we create multiple testable predictions. If the intervention reduces defects but doesn’t improve efficiency, we learn something important about the underlying system mechanics. If it improves efficiency but doesn’t reduce defects, we gain different insights. If it does neither, we discover that our fundamental understanding of the system may be flawed.
This approach requires a cultural shift from celebrating the absence of problems to celebrating the presence of learning. Organizations that embrace falsifiable quality management actively seek conditions that would reveal the limitations of their current systems. They design experiments to test the boundaries of their process capabilities. They view unexpected results not as failures to be explained away, but as opportunities to refine their understanding of system behavior.
The practical implementation of testable risk management involves several key elements:
Hypothesis-Driven Validation: Instead of demonstrating that processes meet specifications, validation activities should test specific hypotheses about process behavior. For example, rather than proving that a sterilization cycle achieves a 6-log reduction, we might test the hypothesis that cycle modifications affect sterility assurance in predictable ways. Instead of demonstrating that the CHO cell culture process consistently produces mAb drug substance meeting predetermined specifications, hypothesis-driven validation would test the specific prediction that maintaining pH at 7.0 ± 0.05 during the production phase will result in final titers that are 15% ± 5% higher than pH maintained at 6.9 ± 0.05, creating a falsifiable hypothesis that can be definitively proven wrong if the predicted titer improvement fails to materialize within the specified confidence intervals
Falsifiable Control Strategies: Control strategies should include specific predictions about how the system will behave under different conditions. These predictions should be testable and potentially falsifiable through routine monitoring or designed experiments.
Learning-Oriented Metrics: Key indicators should be designed to detect when our assumptions about system behavior are incorrect, not just when systems are performing within specification. Metrics that only measure compliance tell us nothing about the underlying system dynamics.
Proactive Stress Testing: Rather than waiting for problems to occur naturally, we should actively probe the boundaries of system performance through controlled stress conditions. This approach reveals failure modes before they impact patients while providing valuable data about system robustness.
Designing Falsifiable Quality Systems
The practical challenge of designing falsifiable quality systems requires a fundamental reconceptualization of how we approach quality assurance. Instead of building systems designed to prevent all possible failures, we need systems designed to fail in instructive ways when our underlying assumptions are incorrect.
This approach starts with making our assumptions explicit and testable. Traditional quality systems often embed numerous unstated assumptions about process behavior, material characteristics, environmental conditions, and human performance. These assumptions are rarely articulated clearly enough to be tested, making the systems inherently unfalsifiable. A falsifiable quality system makes these assumptions explicit and designs tests to evaluate their validity.
Consider the design of a typical pharmaceutical manufacturing process. Traditional approaches focus on demonstrating that the process consistently produces product meeting specifications under defined conditions. This demonstration typically involves process validation studies that show the process works under idealized conditions, followed by ongoing monitoring to detect deviations from expected performance.
A falsifiable approach would start by articulating specific hypotheses about what drives process performance. We might hypothesize that product quality is primarily determined by three critical process parameters, that these parameters interact in predictable ways, and that environmental variations within specified ranges don’t significantly impact these relationships. Each of these hypotheses can be tested and potentially falsified through designed experiments or systematic observation of process performance.
The key insight is that falsifiable quality systems are designed around testable theories of what makes quality systems effective, rather than around defensive strategies for preventing all possible problems. This shift enables genuine learning and continuous improvement because we can distinguish between interventions that work for the right reasons and those that appear to work for unknown or incorrect reasons.
Structured Hypothesis Formation: Quality requirements should be built around explicit hypotheses about cause-and-effect relationships in critical processes. These hypotheses should be specific enough to be tested and potentially falsified through systematic observation or experimentation.
Predictive Monitoring: Instead of monitoring for compliance with specifications, systems should monitor for deviations from predicted behavior. When predictions prove incorrect, this provides valuable information about the accuracy of our underlying process understanding.
Experimental Integration: Routine operations should be designed to provide ongoing tests of system hypotheses. Process changes, material variations, and environmental fluctuations should be treated as natural experiments that provide data about system behavior rather than disturbances to be minimized.
Failure Mode Anticipation: Quality systems should explicitly anticipate the ways failures might happen and design detection mechanisms for these failure modes. This proactive approach contrasts with reactive systems that only detect problems after they occur.
The Evolution of Risk Assessment: From Compliance to Science
The evolution of pharmaceutical risk assessment from compliance-focused activities to genuine scientific inquiry represents one of the most significant opportunities for improving quality outcomes. Traditional risk assessments often function primarily as documentation exercises designed to satisfy regulatory requirements rather than tools for genuine learning and improvement.
ICH Q9(R1) recognizes this limitation and calls for more scientifically rigorous approaches to quality risk management. The updated guidance emphasizes the need for risk assessments to be based on scientific knowledge and to provide actionable insights for quality improvement. This represents a shift away from checklist-based compliance activities toward hypothesis-driven scientific inquiry.
The integration of falsifiability principles with ICH Q9(R1) requirements creates opportunities for more rigorous and useful risk assessments. Instead of asking generic questions about what could go wrong, falsifiable risk assessments develop specific hypotheses about failure modes and design tests to evaluate these hypotheses. This approach provides more actionable insights while meeting regulatory expectations for systematic risk evaluation.
Consider the evolution of Failure Mode and Effects Analysis (FMEA) from a traditional compliance tool to a falsifiable risk assessment method. Traditional FMEA often devolves into generic lists of potential failures with subjective probability and impact assessments. The results provide limited insight because the assessments can’t be systematically tested or validated.
A falsifiable FMEA would start with specific hypotheses about failure mechanisms and their relationships to process parameters, material characteristics, or operational conditions. These hypotheses would be tested through historical data analysis, designed experiments, or systematic monitoring programs. The results would provide genuine insights into system behavior while creating a foundation for continuous improvement.
This evolution requires changes in how we approach several key risk assessment activities:
Hazard Identification: Instead of brainstorming all possible things that could go wrong, risk identification should focus on developing testable hypotheses about specific failure mechanisms and their triggers.
Risk Analysis: Probability and impact assessments should be based on testable models of system behavior rather than subjective expert judgment. When models prove inaccurate, this provides valuable information about the need to revise our understanding of system dynamics.
Risk Control: Control measures should be designed around testable theories of how interventions affect system behavior. The effectiveness of controls should be evaluated through systematic monitoring and periodic testing rather than assumed based on their implementation.
Risk Review: Risk review activities should focus on testing the accuracy of previous risk predictions and updating risk models based on new evidence. This creates a learning loop that continuously improves the quality of risk assessments over time.
Practical Framework for Falsifiable Quality Risk Management
The implementation of falsifiable quality risk management requires a systematic framework that integrates Popperian principles with practical pharmaceutical quality requirements. This framework must be sophisticated enough to generate genuine scientific insights while remaining practical for routine quality management activities.
The foundation of this framework rests on the principle that effective quality systems are built around testable theories of what drives quality outcomes. These theories should make specific predictions that can be evaluated through systematic observation, controlled experimentation, or historical data analysis. When predictions prove incorrect, this provides valuable information about the need to revise our understanding of system behavior.
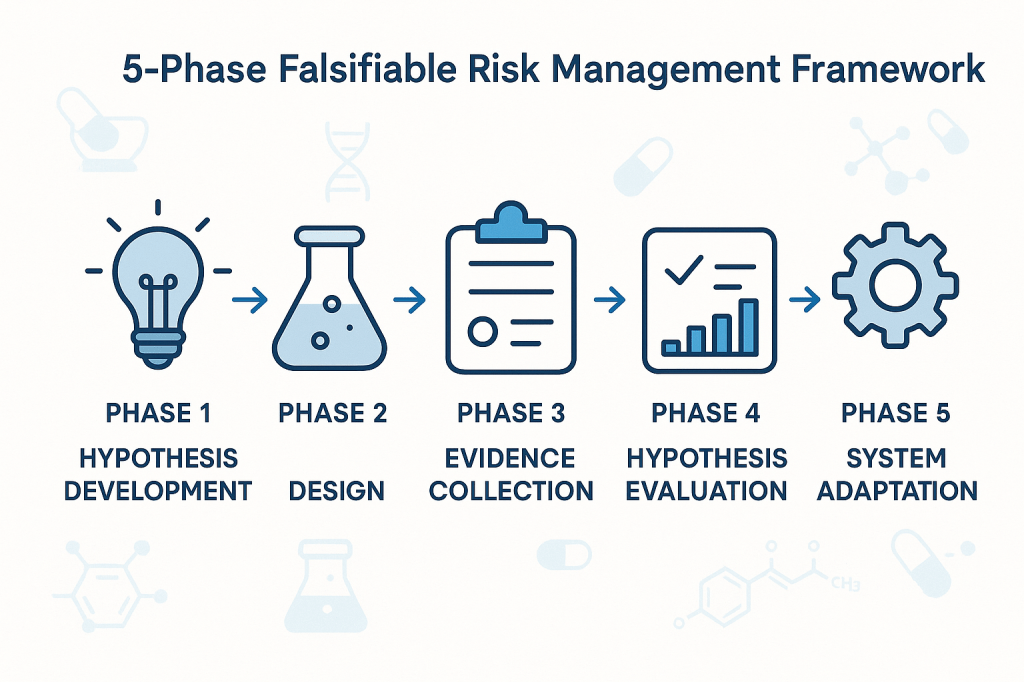
Phase 1: Hypothesis Development
The first phase involves developing specific, testable hypotheses about system behavior. These hypotheses should address fundamental questions about what drives quality outcomes in specific operational contexts. Rather than generic statements about quality risks, hypotheses should make specific predictions about relationships between process parameters, material characteristics, environmental conditions, and quality outcomes.
For example, instead of the generic hypothesis that “temperature variations affect product quality,” a falsifiable hypothesis might state that “temperature excursions above 25°C for more than 30 minutes during the mixing phase increase the probability of out-of-specification results by at least 20%.” This hypothesis is specific enough to be tested and potentially falsified through systematic data collection and analysis.
Phase 2: Experimental Design
The second phase involves designing systematic approaches to test the hypotheses developed in Phase 1. This might involve controlled experiments, systematic analysis of historical data, or structured monitoring programs designed to capture relevant data about hypothesis validity.
The key principle is that testing approaches should be capable of falsifying the hypotheses if they are incorrect. This requires careful attention to statistical power, measurement systems, and potential confounding factors that might obscure true relationships between variables.
Phase 3: Evidence Collection
The third phase focuses on systematic collection of evidence relevant to hypothesis testing. This evidence might come from designed experiments, routine monitoring data, or systematic analysis of historical performance. The critical requirement is that evidence collection should be structured around hypothesis testing rather than generic performance monitoring.
Evidence collection systems should be designed to detect when hypotheses are incorrect, not just when systems are performing within specifications. This requires more sophisticated approaches to data analysis and interpretation than traditional compliance-focused monitoring.
Phase 4: Hypothesis Evaluation
The fourth phase involves systematic evaluation of evidence against the hypotheses developed in Phase 1. This evaluation should follow rigorous statistical methods and should be designed to reach definitive conclusions about hypothesis validity whenever possible.
When hypotheses are falsified, this provides valuable information about the need to revise our understanding of system behavior. When hypotheses are supported by evidence, this provides confidence in our current understanding while suggesting areas for further testing and refinement.
Phase 5: System Adaptation
The final phase involves adapting quality systems based on the insights gained through hypothesis testing. This might involve modifying control strategies, updating risk assessments, or redesigning monitoring programs based on improved understanding of system behavior.
The critical principle is that system adaptations should be based on genuine learning about system behavior rather than reactive responses to compliance issues or external pressures. This creates a foundation for continuous improvement that builds cumulative knowledge about what drives quality outcomes.
Implementation Challenges
The transition to falsifiable quality risk management faces several practical challenges that must be addressed for successful implementation. These challenges range from technical issues related to experimental design and statistical analysis to cultural and organizational barriers that may resist more scientifically rigorous approaches to quality management.
Technical Challenges
The most immediate technical challenge involves designing falsifiable hypotheses that are relevant to pharmaceutical quality management. Many quality professionals have extensive experience with compliance-focused activities but limited experience with experimental design and hypothesis testing. This skills gap must be addressed through targeted training and development programs.
Statistical power represents another significant technical challenge. Many quality systems operate with very low baseline failure rates, making it difficult to design experiments with adequate power to detect meaningful differences in system performance. This requires sophisticated approaches to experimental design and may necessitate longer observation periods or larger sample sizes than traditionally used in quality management.
Measurement systems present additional challenges. Many pharmaceutical quality attributes are difficult to measure precisely, introducing uncertainty that can obscure true relationships between process parameters and quality outcomes. This requires careful attention to measurement system validation and uncertainty quantification.
Cultural and Organizational Challenges
Perhaps more challenging than technical issues are the cultural and organizational barriers to implementing more scientifically rigorous quality management approaches. Many pharmaceutical organizations have deeply embedded cultures that prioritize risk avoidance and compliance over learning and improvement.
The shift to falsifiable quality management requires cultural change that embraces controlled failure as a learning opportunity rather than something to be avoided at all costs. This represents a fundamental change in how many organizations think about quality management and may encounter significant resistance.
Regulatory relationships present additional organizational challenges. Many quality professionals worry that more rigorous scientific approaches to quality management might raise regulatory concerns or create compliance burdens. This requires careful communication with regulatory agencies to demonstrate that falsifiable approaches enhance rather than compromise patient safety.
Strategic Solutions
Successfully implementing falsifiable quality risk management requires strategic approaches that address both technical and cultural challenges. These solutions must be tailored to specific organizational contexts while maintaining scientific rigor and regulatory compliance.
Pilot Programs: Implementation should begin with carefully selected pilot programs in areas where falsifiable approaches can demonstrate clear value. These pilots should be designed to generate success stories that support broader organizational adoption while building internal capability and confidence.
Training and Development: Comprehensive training programs should be developed to build organizational capability in experimental design, statistical analysis, and hypothesis testing. These programs should be tailored to pharmaceutical quality contexts and should emphasize practical applications rather than theoretical concepts.
Regulatory Engagement: Proactive engagement with regulatory agencies should emphasize how falsifiable approaches enhance patient safety through improved understanding of system behavior. This communication should focus on the scientific rigor of the approach rather than on business benefits that might appear secondary to regulatory objectives.
Cultural Change Management: Systematic change management programs should address cultural barriers to embracing controlled failure as a learning opportunity. These programs should emphasize how falsifiable approaches support regulatory compliance and patient safety rather than replacing these priorities with business objectives.
Case Studies: Falsifiability in Practice
The practical application of falsifiable quality risk management can be illustrated through several case studies that demonstrate how Popperian principles can be integrated with routine pharmaceutical quality activities. These examples show how hypotheses can be developed, tested, and used to improve quality outcomes while maintaining regulatory compliance.
Case Study 1: Cleaning Validation Optimization
A biologics manufacturer was experiencing occasional cross-contamination events despite having validated cleaning procedures that consistently met acceptance criteria. Traditional approaches focused on demonstrating that cleaning procedures reduced contamination below specified limits, but provided no insight into the factors that occasionally caused this system to fail.
The falsifiable approach began with developing specific hypotheses about cleaning effectiveness. The team hypothesized that cleaning effectiveness was primarily determined by three factors: contact time with cleaning solution, mechanical action intensity, and rinse water temperature. They further hypothesized that these factors interacted in predictable ways and that current procedures provided a specific margin of safety above minimum requirements.
These hypotheses were tested through a designed experiment that systematically varied each cleaning parameter while measuring residual contamination levels. The results revealed that current procedures were adequate under ideal conditions but provided minimal margin of safety when multiple factors were simultaneously at their worst-case levels within specified ranges.
Based on these findings, the cleaning procedure was modified to provide greater margin of safety during worst-case conditions. More importantly, ongoing monitoring was redesigned to test the continued validity of the hypotheses about cleaning effectiveness rather than simply verifying compliance with acceptance criteria.
Case Study 2: Process Control Strategy Development
A pharmaceutical manufacturer was developing a control strategy for a new manufacturing process. Traditional approaches would have focused on identifying critical process parameters and establishing control limits based on process validation studies. Instead, the team used a falsifiable approach that started with explicit hypotheses about process behavior.
The team hypothesized that product quality was primarily controlled by the interaction between temperature and pH during the reaction phase, that these parameters had linear effects on product quality within the normal operating range, and that environmental factors had negligible impact on these relationships.
These hypotheses were tested through systematic experimentation during process development. The results confirmed the importance of the temperature-pH interaction but revealed nonlinear effects that weren’t captured in the original hypotheses. More importantly, environmental humidity was found to have significant effects on process behavior under certain conditions.
The control strategy was designed around the revised understanding of process behavior gained through hypothesis testing. Ongoing process monitoring was structured to continue testing key assumptions about process behavior rather than simply detecting deviations from target conditions.
Case Study 3: Supplier Quality Management
A biotechnology company was managing quality risks from a critical raw material supplier. Traditional approaches focused on incoming inspection and supplier auditing to verify compliance with specifications and quality system requirements. However, occasional quality issues suggested that these approaches weren’t capturing all relevant quality risks.
The falsifiable approach started with specific hypotheses about what drove supplier quality performance. The team hypothesized that supplier quality was primarily determined by their process control during critical manufacturing steps, that certain environmental conditions increased the probability of quality issues, and that supplier quality system maturity was predictive of long-term quality performance.
These hypotheses were tested through systematic analysis of supplier quality data, enhanced supplier auditing focused on specific process control elements, and structured data collection about environmental conditions during material manufacturing. The results revealed that traditional quality system assessments were poor predictors of actual quality performance, but that specific process control practices were strongly predictive of quality outcomes.
The supplier management program was redesigned around the insights gained through hypothesis testing. Instead of generic quality system requirements, the program focused on specific process control elements that were demonstrated to drive quality outcomes. Supplier performance monitoring was structured around testing continued validity of the relationships between process control and quality outcomes.
Measuring Success in Falsifiable Quality Systems
The evaluation of falsifiable quality systems requires fundamentally different approaches to performance measurement than traditional compliance-focused systems. Instead of measuring the absence of problems, we need to measure the presence of learning and the accuracy of our predictions about system behavior.
Traditional quality metrics focus on outcomes: defect rates, deviation frequencies, audit findings, and regulatory observations. While these metrics remain important for regulatory compliance and business performance, they provide limited insight into whether our quality systems are actually effective or merely lucky. Falsifiable quality systems require additional metrics that evaluate the scientific validity of our approach to quality management.
Predictive Accuracy Metrics
The most direct measure of a falsifiable quality system’s effectiveness is the accuracy of its predictions about system behavior. These metrics evaluate how well our hypotheses about quality system behavior match observed outcomes. High predictive accuracy suggests that we understand the underlying drivers of quality outcomes. Low predictive accuracy indicates that our understanding needs refinement.
Predictive accuracy metrics might include the percentage of process control predictions that prove correct, the accuracy of risk assessments in predicting actual quality issues, or the correlation between predicted and observed responses to process changes. These metrics provide direct feedback about the validity of our theoretical understanding of quality systems.
Learning Rate Metrics
Another important category of metrics evaluates how quickly our understanding of quality systems improves over time. These metrics measure the rate at which falsified hypotheses lead to improved system performance or more accurate predictions. High learning rates indicate that the organization is effectively using falsifiable approaches to improve quality outcomes.
Learning rate metrics might include the time required to identify and correct false assumptions about system behavior, the frequency of successful process improvements based on hypothesis testing, or the rate of improvement in predictive accuracy over time. These metrics evaluate the dynamic effectiveness of falsifiable quality management approaches.
Hypothesis Quality Metrics
The quality of hypotheses generated by quality risk management processes represents another important performance dimension. High-quality hypotheses are specific, testable, and relevant to important quality outcomes. Poor-quality hypotheses are vague, untestable, or focused on trivial aspects of system performance.
Hypothesis quality can be evaluated through structured peer review processes, assessment of testability and specificity, and evaluation of relevance to critical quality attributes. Organizations with high-quality hypothesis generation processes are more likely to gain meaningful insights from their quality risk management activities.
System Robustness Metrics
Falsifiable quality systems should become more robust over time as learning accumulates and system understanding improves. Robustness can be measured through the system’s ability to maintain performance despite variations in operating conditions, changes in materials or equipment, or other sources of uncertainty.
Robustness metrics might include the stability of process performance across different operating conditions, the effectiveness of control strategies under stress conditions, or the system’s ability to detect and respond to emerging quality risks. These metrics evaluate whether falsifiable approaches actually lead to more reliable quality systems.
Regulatory Implications and Opportunities
The integration of falsifiable principles with pharmaceutical quality risk management creates both challenges and opportunities in regulatory relationships. While some regulatory agencies may initially view scientific approaches to quality management with skepticism, the ultimate result should be enhanced regulatory confidence in quality systems that can demonstrate genuine understanding of what drives quality outcomes.
The key to successful regulatory engagement lies in emphasizing how falsifiable approaches enhance patient safety rather than replacing regulatory compliance with business optimization. Regulatory agencies are primarily concerned with patient safety and product quality. Falsifiable quality systems support these objectives by providing more rigorous and reliable approaches to ensuring quality outcomes.
Enhanced Regulatory Submissions
Regulatory submissions based on falsifiable quality systems can provide more compelling evidence of system effectiveness than traditional compliance-focused approaches. Instead of demonstrating that systems meet minimum requirements, falsifiable approaches can show genuine understanding of what drives quality outcomes and how systems will behave under different conditions.
This enhanced evidence can support regulatory flexibility in areas such as process validation, change control, and ongoing monitoring requirements. Regulatory agencies may be willing to accept risk-based approaches to these activities when they’re supported by rigorous scientific evidence rather than generic compliance activities.
Proactive Risk Communication
Falsifiable quality systems enable more proactive and meaningful communication with regulatory agencies about quality risks and mitigation strategies. Instead of reactive communication about compliance issues, organizations can engage in scientific discussions about system behavior and improvement strategies.
This proactive communication can build regulatory confidence in organizational quality management capabilities while providing opportunities for regulatory agencies to provide input on scientific approaches to quality improvement. The result should be more collaborative regulatory relationships based on shared commitment to scientific rigor and patient safety.
Regulatory Science Advancement
The pharmaceutical industry’s adoption of more scientifically rigorous approaches to quality management can contribute to the advancement of regulatory science more broadly. Regulatory agencies benefit from industry innovations in risk assessment, process understanding, and quality assurance methods.
Organizations that successfully implement falsifiable quality risk management can serve as case studies for regulatory guidance development and can provide evidence for the effectiveness of science-based approaches to quality assurance. This contribution to regulatory science advancement creates value that extends beyond individual organizational benefits.
Toward a More Scientific Quality Culture
The long-term vision for falsifiable quality risk management extends beyond individual organizational implementations to encompass fundamental changes in how the pharmaceutical industry approaches quality assurance. This vision includes more rigorous scientific approaches to quality management, enhanced collaboration between industry and regulatory agencies, and continuous advancement in our understanding of what drives quality outcomes.
Industry-Wide Learning Networks
One promising direction involves the development of industry-wide learning networks that share insights from falsifiable quality management implementations. These networks facilitate collaborative hypothesis testing, shared learning from experimental results, and development of common methodologies for scientific approaches to quality assurance.
Such networks accelerate the advancement of quality science while maintaining appropriate competitive boundaries. Organizations should share methodological insights and general findings while protecting proprietary information about specific processes or products. The result would be faster advancement in quality management science that benefits the entire industry.
Advanced Analytics Integration
The integration of advanced analytics and machine learning techniques with falsifiable quality management approaches represents another promising direction. These technologies can enhance our ability to develop testable hypotheses, design efficient experiments, and analyze complex datasets to evaluate hypothesis validity.
Machine learning approaches are particularly valuable for identifying patterns in complex quality datasets that might not be apparent through traditional analysis methods. However, these approaches must be integrated with falsifiable frameworks to ensure that insights can be validated and that predictive models can be systematically tested and improved.
Regulatory Harmonization
The global harmonization of regulatory approaches to science-based quality management represents a significant opportunity for advancing patient safety and regulatory efficiency. As individual regulatory agencies gain experience with falsifiable quality management approaches, there are opportunities to develop harmonized guidance that supports consistent global implementation.
ICH Q9(r1) was a great step. I would love to see continued work in this area.
Embracing the Discomfort of Scientific Rigor
The transition from compliance-focused to scientifically rigorous quality risk management represents more than a methodological change—it requires fundamentally rethinking how we approach quality assurance in pharmaceutical manufacturing. By embracing Popper’s challenge that genuine scientific theories must be falsifiable, we move beyond the comfortable but ultimately unhelpful world of proving negatives toward the more demanding but ultimately more rewarding world of testing positive claims about system behavior.
The effectiveness paradox that motivates this discussion—the problem of determining what works when our primary evidence is that “nothing bad happened”—cannot be resolved through better compliance strategies or more sophisticated documentation. It requires genuine scientific inquiry into the mechanisms that drive quality outcomes. This inquiry must be built around testable hypotheses that can be proven wrong, not around defensive strategies that can always accommodate any possible outcome.
The practical implementation of falsifiable quality risk management is not without challenges. It requires new skills, different cultural approaches, and more sophisticated methodologies than traditional compliance-focused activities. However, the potential benefits—genuine learning about system behavior, more reliable quality outcomes, and enhanced regulatory confidence—justify the investment required for successful implementation.
Perhaps most importantly, the shift to falsifiable quality management moves us toward a more honest assessment of what we actually know about quality systems versus what we merely assume or hope to be true. This honesty is uncomfortable but essential for building quality systems that genuinely serve patient safety rather than organizational comfort.
The question is not whether pharmaceutical quality management will eventually embrace more scientific approaches—the pressures of regulatory evolution, competitive dynamics, and patient safety demands make this inevitable. The question is whether individual organizations will lead this transition or be forced to follow. Those that embrace the discomfort of scientific rigor now will be better positioned to thrive in a future where quality management is evaluated based on genuine effectiveness rather than compliance theater.
As we continue to navigate an increasingly complex regulatory and competitive environment, the organizations that master the art of turning uncertainty into testable knowledge will be best positioned to deliver consistent quality outcomes while maintaining the flexibility needed for innovation and continuous improvement. The integration of Popperian falsifiability with modern quality risk management provides a roadmap for achieving this mastery while maintaining the rigorous standards our industry demands.
The path forward requires courage to question our current assumptions, discipline to design rigorous tests of our theories, and wisdom to learn from both our successes and our failures. But for those willing to embrace these challenges, the reward is quality systems that are not only compliant but genuinely effective. Systems that we can defend not because they’ve never been proven wrong, but because they’ve been proven right through systematic, scientific inquiry.
The pharmaceutical industry has operated for over a decade under the comfortable assumption that GAMP 5’s risk-based guidance for system requirements represented industry best practice—helpful, comprehensive, but ultimately voluntary. Section 6 of the draft Annex 11 moves many things from recommended to mandated. What GAMP 5 suggested as scalable guidance, Annex 11 codifies as enforceable regulation. For computer system validation professionals, this isn’t just an update—it’s a fundamental shift from “how we should do it” to “how we must do it.”
This transformation carries profound implications that extend far beyond documentation requirements. Section 6 represents the regulatory codification of modern system engineering practices, forcing organizations to abandon the shortcuts, compromises, and “good enough” approaches that have persisted despite GAMP 5’s guidance. More significantly, it establishes system requirements as the immutable foundation of validation rather than merely an input to the process.
For CSV experts who have spent years evangelizing GAMP 5 principles within organizations that treated requirements as optional documentation, Section 6 provides regulatory teeth that will finally compel comprehensive implementation. However, it also raises the stakes dramatically—what was once best practice guidance subject to interpretation becomes regulatory obligation subject to inspection.
The Mandatory Transformation: From Guidance to Regulation
6.1: GMP Functionality—The End of Requirements Optionality
The opening requirement of Section 6 eliminates any ambiguity about system requirements documentation: “A regulated user should establish and approve a set of system requirements (e.g. a User Requirements Specification, URS), which accurately describe the functionality the regulated user has automated and is relying on when performing GMP activities.”
This language transforms what GAMP 5 positioned as risk-based guidance into regulatory mandate. The phrase “should establish and approve” in regulatory context carries the force of “must“—there is no longer discretion about whether to document system requirements. Every computerized system touching GMP activities requires formal requirements documentation, regardless of system complexity, development approach, or organizational preference.
The scope is deliberately comprehensive, explicitly covering “whether a system is developed in-house, is a commercial off-the-shelf product, or is provided as-a-service” and “independently on whether it is developed following a linear or iterative software development process.” This eliminates common industry escapes: cloud services can’t claim exemption because they’re external; agile development can’t avoid documentation because it’s iterative; COTS systems can’t rely solely on vendor documentation because they’re pre-built.
The requirement for accuracy in describing “functionality the regulated user has automated and is relying on” establishes a direct link between system capabilities and GMP dependencies. Organizations must explicitly identify and document what GMP activities depend on system functionality, creating traceability between business processes and technical capabilities that many current validation approaches lack.
Major Strike Against the Concept of “Indirect”
The new draft Annex 11 explicitly broadens the scope of requirements for user requirements specifications (URS) and validation to cover all computerized systems with GMP relevance—not just those with direct product or decision-making impact, but also indirect GMP systems. This means systems that play a supporting or enabling role in GMP activities (such as underlying IT infrastructure, databases, cloud services, SaaS platforms, integrated interfaces, and any outsourced or vendor-managed digital environments) are fully in scope.
Section 6 of the draft states that user requirements must “accurately describe the functionality the regulated user has automated and is relying on when performing GMP activities,” with no exemption or narrower definition for indirect systems. It emphasizes that this principle applies “regardless of whether a system is developed in-house, is a commercial off-the-shelf product, or is provided as-a-service, and independently of whether it is developed following a linear or iterative software development process.” The regulated user is responsible for approving, controlling, and maintaining these requirements over the system’s lifecycle—even if the system is managed by a third party or only indirectly involved in GMP data or decision workflows.
Importantly, the language and supporting commentaries make it clear that traceability of user requirements throughout the lifecycle is mandatory for all systems with GMP impact—direct or indirect. There is no explicit exemption in the draft for indirect GMP systems. Regulatory and industry analyses confirm that the burden of documented, risk-assessed, and lifecycle-maintained user requirements sits equally with indirect systems as with direct ones, as long as they play a role in assuring product quality, patient safety, or data integrity.
In practice, this means organizations must extend their URS, specification, and validation controls to any computerized system that through integration, support, or data processing could influence GMP compliance. The regulated company remains responsible for oversight, traceability, and quality management of those systems, whether or not they are operated by a vendor or IT provider. This is a significant expansion from previous regulatory expectations and must be factored into computerized system inventories, risk assessments, and validation strategies going forward.
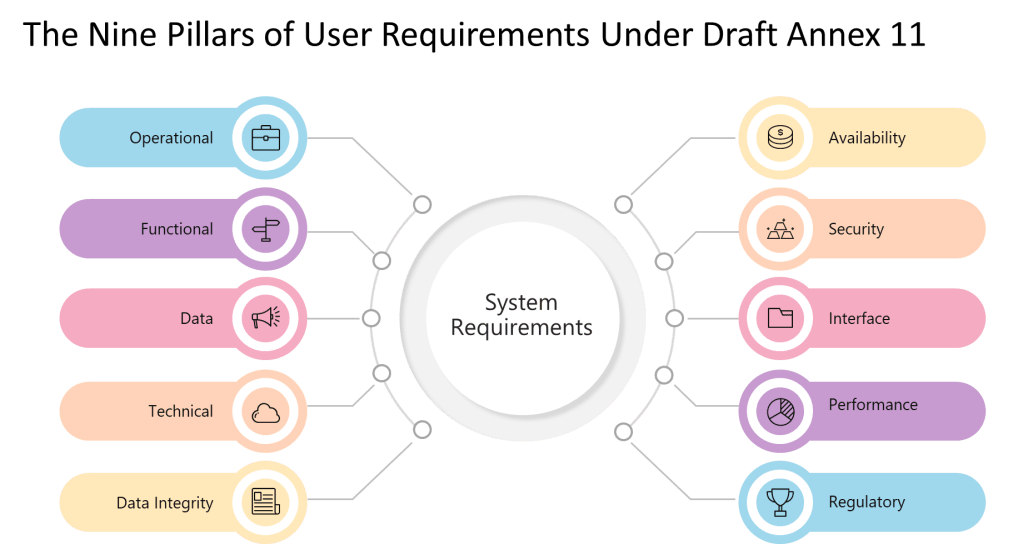
9 Pillars of a User Requirements
Pillar
Description
Practical Examples
Operational
Requirements describing how users will operate the system for GMP tasks.
Workflow steps, user roles, batch record creation.
Functional
Features and functions the system must perform to support GMP processes.
Explicit requirements imposed by GMP regulations and standards.
Part 11/Annex 11 compliance, data retention, auditability.
6.2: Extent and Detail—Risk-Based Rigor, Not Risk-Based Avoidance
Section 6.2 appears to maintain GAMP 5’s risk-based philosophy by requiring that “extent and detail of defined requirements should be commensurate with the risk, complexity and novelty of a system.” However, the subsequent specifications reveal a much more prescriptive approach than traditional risk-based frameworks.
The requirement that descriptions be “sufficient to support subsequent risk analysis, specification, design, purchase, configuration, qualification and validation” establishes requirements documentation as the foundation for the entire system lifecycle. This moves beyond GAMP 5’s emphasis on requirements as input to validation toward positioning requirements as the definitive specification against which all downstream activities are measured.
The explicit enumeration of requirement types—”operational, functional, data integrity, technical, interface, performance, availability, security, and regulatory requirements”—represents a significant departure from GAMP 5’s more flexible categorization. Where GAMP 5 allows organizations to define requirement categories based on system characteristics and business needs, Annex 11 mandates coverage of nine specific areas regardless of system type or risk level.
This prescriptive approach reflects regulatory recognition that organizations have historically used “risk-based” as justification for inadequate requirements documentation. By specifying minimum coverage areas, Section 6 establishes a floor below which requirements documentation cannot fall, regardless of risk assessment outcomes.
The inclusion of “process maps and data flow diagrams” as recommended content acknowledges the reality that modern pharmaceutical operations involve complex, interconnected systems where understanding data flows and process dependencies is essential for effective validation. This requirement will force organizations to develop system-level understanding rather than treating validation as isolated technical testing.
6.3: Ownership—User Accountability in the Cloud Era
Perhaps the most significant departure from traditional industry practice, Section 6.3 addresses the growing trend toward cloud services and vendor-supplied systems by establishing unambiguous user accountability for requirements documentation. The requirement that “the regulated user should take ownership of the document covering the implemented version of the system and formally approve and control it” eliminates common practices where organizations rely entirely on vendor-provided documentation.
This requirement acknowledges that vendor-supplied requirements specifications rarely align perfectly with specific organizational needs, GMP processes, or regulatory expectations. While vendors may provide generic requirements documentation suitable for broad market applications, pharmaceutical organizations must customize, supplement, and formally adopt these requirements to reflect their specific implementation and GMP dependencies.
The language “carefully review and approve the document and consider whether the system fulfils GMP requirements and company processes as is, or whether it should be configured or customised” requires active evaluation rather than passive acceptance. Organizations cannot simply accept vendor documentation as sufficient—they must demonstrate that they have evaluated system capabilities against their specific GMP needs and either confirmed alignment or documented necessary modifications.
This ownership requirement will prove challenging for organizations using large cloud platforms or SaaS solutions where vendors resist customization of standard documentation. However, the regulatory expectation is clear: pharmaceutical companies cannot outsource responsibility for demonstrating that system capabilities meet their specific GMP requirements.
6.4: Update—Living Documentation, Not Static Archives
Section 6.4 addresses one of the most persistent failures in current validation practice: requirements documentation that becomes obsolete immediately after initial validation. The requirement that “requirements should be updated and maintained throughout the lifecycle of a system” and that “updated requirements should form the very basis for qualification and validation” establishes requirements as living documentation rather than historical artifacts.
This approach reflects the reality that modern computerized systems undergo continuous change through software updates, configuration modifications, hardware refreshes, and process improvements. Traditional validation approaches that treat requirements as fixed specifications become increasingly disconnected from operational reality as systems evolve.
The phrase “form the very basis for qualification and validation” positions requirements documentation as the definitive specification against which system performance is measured throughout the lifecycle. This means that any system change must be evaluated against current requirements, and any requirements change must trigger appropriate validation activities.
This requirement will force organizations to establish requirements management processes that rival those used in traditional software development organizations. Requirements changes must be controlled, evaluated for impact, and reflected in validation documentation—capabilities that many pharmaceutical organizations currently lack.
6.5: Traceability—Engineering Discipline for Validation
The traceability requirement in Section 6.5 codifies what GAMP 5 has long recommended: “Documented traceability between individual requirements, underlaying design specifications and corresponding qualification and validation test cases should be established and maintained.” However, the regulatory context transforms this from validation best practice to compliance obligation.
The emphasis on “effective tools to capture and hold requirements and facilitate the traceability” acknowledges that manual traceability management becomes impractical for complex systems with hundreds or thousands of requirements. This requirement will drive adoption of requirements management tools and validation platforms that can maintain automated traceability throughout the system lifecycle.
Traceability serves multiple purposes in the validation context: ensuring comprehensive test coverage, supporting impact assessment for changes, and providing evidence of validation completeness. Section 6 positions traceability as fundamental validation infrastructure rather than optional documentation enhancement.
For organizations accustomed to simplified validation approaches where test cases are developed independently of detailed requirements, this traceability requirement represents a significant process change requiring tool investment and training.
6.6: Configuration—Separating Standard from Custom
The final subsection addresses configuration management by requiring clear documentation of “what functionality, if any, is modified or added by configuration of a system.” This requirement recognizes that most modern pharmaceutical systems involve significant configuration rather than custom development, and that configuration decisions have direct impact on validation scope and approaches.
The distinction between standard system functionality and configured functionality is crucial for validation planning. Standard functionality may be covered by vendor testing and certification, while configured functionality requires user validation. Section 6 requires this distinction to be explicit and documented.
The requirement for “controlled configuration specification” separate from requirements documentation reflects recognition that configuration details require different management approaches than functional requirements. Configuration specifications must reflect the actual system implementation rather than desired capabilities.
Comparison with GAMP 5: Evolution Becomes Revolution
Philosophical Alignment with Practical Divergence
Section 6 maintains GAMP 5’s fundamental philosophy—risk-based validation supported by comprehensive requirements documentation—while dramatically changing implementation expectations. Both frameworks emphasize user ownership of requirements, lifecycle management, and traceability as essential validation elements. However, the regulatory context of Annex 11 transforms voluntary guidance into enforceable obligation.
GAMP 5’s flexibility in requirements categorization and documentation approaches reflects its role as guidance suitable for diverse organizational contexts and system types. Section 6’s prescriptive approach reflects regulatory recognition that flexibility has often been interpreted as optionality, leading to inadequate requirements documentation that fails to support effective validation.
The risk-based approach remains central to both frameworks, but Section 6 establishes minimum standards that apply regardless of risk assessment outcomes. While GAMP 5 might suggest that low-risk systems require minimal requirements documentation, Section 6 mandates coverage of nine requirement areas for all GMP systems.
Documentation Structure and Content
GAMP 5’s traditional document hierarchy—URS, Functional Specification, Design Specification—becomes more fluid under Section 6, which focuses on ensuring comprehensive coverage rather than prescribing specific document structures. This reflects recognition that modern development approaches, including agile and DevOps practices, may not align with traditional waterfall documentation models.
However, Section 6’s explicit enumeration of requirement types provides more prescriptive guidance than GAMP 5’s flexible approach. Where GAMP 5 might allow organizations to define requirement categories based on system characteristics, Section 6 mandates coverage of operational, functional, data integrity, technical, interface, performance, availability, security, and regulatory requirements.
The emphasis on process maps, data flow diagrams, and use cases reflects modern system complexity where understanding interactions and dependencies is essential for effective validation. GAMP 5 recommends these approaches for complex systems; Section 6 suggests their use “where relevant” for all systems.
Vendor and Service Provider Management
Both frameworks emphasize user responsibility for requirements even when vendors provide initial documentation. However, Section 6 uses stronger language about user ownership and control, reflecting increased regulatory concern about organizations that delegate requirements definition to vendors without adequate oversight.
GAMP 5’s guidance on supplier assessment and leveraging vendor documentation remains relevant under Section 6, but the regulatory requirement for user ownership and approval creates higher barriers for simply accepting vendor-provided documentation as sufficient.
Implementation Challenges for CSV Professionals
Organizational Capability Development
Most pharmaceutical organizations will require significant capability development to meet Section 6 requirements effectively. Traditional validation teams focused on testing and documentation must develop requirements engineering capabilities comparable to those found in software development organizations.
This transformation requires investment in requirements management tools, training for validation professionals, and establishment of requirements governance processes. Organizations must develop capabilities for requirements elicitation, analysis, specification, validation, and change management throughout the system lifecycle.
The traceability requirement particularly challenges organizations accustomed to informal relationships between requirements and test cases. Automated traceability management requires tool investments and process changes that many validation teams are unprepared to implement.
Integration with Existing Validation Approaches
Section 6 requirements must be integrated with existing validation methodologies and documentation structures. Organizations following traditional IQ/OQ/PQ approaches must ensure that requirements documentation supports and guides qualification activities rather than existing as parallel documentation.
The requirement for requirements to “form the very basis for qualification and validation” means that test cases must be explicitly derived from and traceable to documented requirements. This may require significant changes to existing qualification protocols and test scripts.
Organizations using risk-based validation approaches aligned with GAMP 5 guidance will find philosophical alignment with Section 6 but must adapt to more prescriptive requirements for documentation content and structure.
Technology and Tool Requirements
Effective implementation of Section 6 requirements typically requires requirements management tools capable of supporting specification, traceability, change control, and lifecycle management. Many pharmaceutical validation teams currently lack access to such tools or experience in their use.
Tool selection must consider integration with existing validation platforms, support for regulated environments, and capabilities for automated traceability maintenance. Organizations may need to invest in new validation platforms or significantly upgrade existing capabilities.
The emphasis on maintaining requirements throughout the system lifecycle requires tools that support ongoing requirements management rather than just initial documentation. This may conflict with validation approaches that treat requirements as static inputs to qualification activities.
Strategic Implications for the Industry
Convergence of Software Engineering and Pharmaceutical Validation
Section 6 represents convergence between pharmaceutical validation practices and mainstream software engineering approaches. Requirements engineering, long established in software development, becomes mandatory for pharmaceutical computerized systems regardless of development approach or vendor involvement.
This convergence benefits the industry by leveraging proven practices from software engineering while maintaining the rigor and documentation requirements essential for regulated environments. However, it requires pharmaceutical organizations to develop capabilities traditionally associated with software development rather than manufacturing and quality assurance.
The result should be more robust validation practices better aligned with modern system development approaches and capable of supporting the complex, interconnected systems that characterize contemporary pharmaceutical operations.
Vendor Relationship Evolution
Section 6 requirements will reshape relationships between pharmaceutical companies and system vendors. The requirement for user ownership of requirements documentation means that vendors must support more sophisticated requirements management processes rather than simply providing generic specifications.
Vendors that can demonstrate alignment with Section 6 requirements through comprehensive documentation, traceability tools, and support for user customization will gain competitive advantages. Those that resist pharmaceutical-specific requirements management approaches may find their market opportunities limited.
The emphasis on configuration management will drive vendors to provide clearer distinctions between standard functionality and customer-specific configurations, supporting more effective validation planning and execution.
The Regulatory Codification of Modern Validation
Section 6 of the draft Annex 11 represents the regulatory codification of modern computerized system validation practices. What GAMP 5 recommended through guidance, Annex 11 mandates through regulation. What was optional becomes obligatory; what was flexible becomes prescriptive; what was best practice becomes compliance requirement.
For CSV professionals, Section 6 provides regulatory support for comprehensive validation approaches while raising the stakes for inadequate implementation. Organizations that have struggled to implement effective requirements management now face regulatory obligation rather than just professional guidance.
The transformation from guidance to regulation eliminates organizational discretion about requirements documentation quality and comprehensiveness. While risk-based approaches remain valid for scaling validation effort, minimum standards now apply regardless of risk assessment outcomes.
Success under Section 6 requires pharmaceutical organizations to embrace software engineering practices for requirements management while maintaining the documentation rigor and process control essential for regulated environments. This convergence benefits the industry by improving validation effectiveness while ensuring compliance with evolving regulatory expectations.
The industry faces a choice: proactively develop capabilities to meet Section 6 requirements or reactively respond to inspection findings and enforcement actions. For organizations serious about digital transformation and validation excellence, Section 6 provides a roadmap for regulatory-compliant modernization of validation practices.
Requirement Area
Draft Annex 11 Section 6
GAMP 5 Requirements
Key Implementation Considerations
System Requirements Documentation
Mandatory – Must establish and approve system requirements (URS)
Recommended – URS should be developed based on system category and complexity
Organizations must document requirements for ALL GMP systems, regardless of size or complexity
Risk-Based Approach
Extent and detail must be commensurate with risk, complexity, and novelty
Risk-based approach fundamental – validation effort scaled to risk
Risk assessment determines documentation detail but cannot eliminate requirement categories
Functional Requirements
Must include 9 specific requirement types: operational, functional, data integrity, technical, interface, performance, availability, security, regulatory
Functional requirements should be SMART (Specific, Measurable, Achievable, Realistic, Testable)
All 9 areas must be addressed; risk determines depth, not coverage
Traceability Requirements
Documented traceability between requirements, design specs, and test cases required
Traceability matrix recommended – requirements linked through design to testing
Requires investment in traceability tools and processes for complex systems
Requirement Ownership
Regulated user must take ownership even if vendor provides initial requirements
User ownership emphasized, even for purchased systems
Cannot simply accept vendor documentation; must customize and formally approve
Lifecycle Management
Requirements must be updated and maintained throughout system lifecycle
Requirements managed through change control throughout lifecycle
Requires ongoing requirements management process, not just initial documentation
Configuration Management
Configuration options must be described in requirements; chosen configuration documented in controlled spec
Configuration specifications separate from URS
Must clearly distinguish between standard functionality and configured features
Vendor-Supplied Requirements
Vendor requirements must be reviewed, approved, and owned by regulated user
Supplier assessment required – leverage supplier documentation where appropriate
Higher burden on users to customize vendor documentation for specific GMP needs
Validation Basis
Updated requirements must form basis for system qualification and validation
Requirements drive validation strategy and testing scope
Requirements become definitive specification against which system performance is measured
The concept of emergence—where complex behaviors arise unpredictably from interactions among simpler components—has haunted and inspired quality professionals since Aristotle first observed that “the whole is something besides the parts.” In modern quality systems, this ancient paradox takes new form: our meticulously engineered controls often birth unintended consequences, from phantom batch failures to self-reinforcing compliance gaps. Understanding emergence isn’t just an academic exercise—it’s a survival skill in an era where hyperconnected processes and globalized supply chains amplify systemic unpredictability.
The Spectrum of Emergence: From Predictable to Baffling
Emergence manifests across a continuum of complexity, each type demanding distinct management approaches:
1. Simple Emergence Predictable patterns emerge from component interactions, observable even in abstracted models. Consider document control workflows: while individual steps like review or approval seem straightforward, their sequencing creates emergent properties like approval cycle times. These can be precisely modeled using flowcharts or digital twins, allowing proactive optimization.
2. Weak Emergence Behaviors become explainable only after they occur, requiring detailed post-hoc analysis. A pharmaceutical company’s CAPA system might show seasonal trends in effectiveness—a pattern invisible in individual case reviews but emerging from interactions between manufacturing schedules, audit cycles, and supplier quality fluctuations. Weak emergence often reveals itself through advanced analytics like machine learning clustering.
3. Multiple Emergence Here, system behaviors directly contradict component properties. A validated sterile filling line passing all IQ/OQ/PQ protocols might still produce unpredictable media fill failures when integrated with warehouse scheduling software. This “emergent invalidation” stems from hidden interaction vectors that only manifest at full operational scale.
4. Strong Emergence Consistent with components but unpredictably manifested, strong emergence plagues culture-driven quality systems. A manufacturer might implement identical training programs across global sites, yet some facilities develop proactive quality innovation while others foster blame-avoidance rituals. The difference emerges from subtle interactions between local leadership styles and corporate KPIs.
5. Spooky Emergence The most perplexing category, where system behaviors defy both component properties and simulation. A medical device company once faced identical cleanrooms producing statistically divergent particulate counts—despite matching designs, procedures, and personnel. Root cause analysis eventually traced the emergence to nanometer-level differences in HVAC duct machining, interacting with shift-change lighting schedules to alter airflow dynamics.
Type
Characteristics
Quality System Example
Simple
Predictable through component analysis
Document control workflows
Weak
Explainable post-occurrence through detailed modeling
Consistent with components but unpredictably manifested
Culture-driven quality behaviors
Spooky
Defies component properties and simulation entirely
Phantom batch failures in identical systems
The Modern Catalysts of Emergence
Three forces amplify emergence in contemporary quality systems:
Hyperconnected Processes
IoT-enabled manufacturing equipment generates real-time data avalanches. A biologics plant’s environmental monitoring system might integrate 5,000 sensors updating every 15 seconds. The emergent property? A “data tide” that overwhelms traditional statistical process control, requiring AI-driven anomaly detection to discern meaningful signals.
Compressed Innovation Cycles
Compressed innovation cycles are transforming the landscape of product development and quality management. In this new paradigm, the pressure to deliver products faster—whether due to market demands, technological advances, or public health emergencies—means that the traditional, sequential approach to development is replaced by a model where multiple phases run in parallel. Design, manufacturing, and validation activities that once followed a linear path now overlap, requiring organizations to verify quality in real time rather than relying on staged reviews and lengthy data collection.
One of the most significant consequences of this acceleration is the telescoping of validation windows. Where stability studies and shelf-life determinations once spanned years, they are now compressed into a matter of months or even weeks. This forces quality teams to make critical decisions based on limited data, often relying on predictive modeling and statistical extrapolation to fill in the gaps. The result is what some call “validation debt”—a situation where the pace of development outstrips the accumulation of empirical evidence, leaving organizations to manage risks that may not be fully understood until after product launch.
Regulatory frameworks are also evolving in response to compressed innovation cycles. Instead of the traditional, comprehensive submission and review process, regulators are increasingly open to iterative, rolling reviews and provisional specifications that can be adjusted as more data becomes available post-launch. This shift places greater emphasis on computational evidence, such as in silico modeling and digital twins, rather than solely on physical testing and historical precedent.
The acceleration of development timelines amplifies the risk of emergent behaviors within quality systems. Temporal compression means that components and subsystems are often scaled up and integrated before they have been fully characterized or validated in isolation. This can lead to unforeseen interactions and incompatibilities that only become apparent at the system level, sometimes after the product has reached the market. The sheer volume and velocity of data generated in these environments can overwhelm traditional quality monitoring tools, making it difficult to identify and respond to critical quality attributes in a timely manner.
Another challenge arises from the collision of different quality management protocols. As organizations attempt to blend frameworks such as GMP, Agile, and Lean to keep pace with rapid development, inconsistencies and gaps can emerge. Cross-functional teams may interpret standards differently, leading to confusion or conflicting priorities that undermine the integrity of the quality system.
The systemic consequences of compressed innovation cycles are profound. Cryptic interaction pathways can develop, where components that performed flawlessly in isolation begin to interact in unexpected ways at scale. Validation artifacts—such as artificial stability observed in accelerated testing—may fail to predict real-world performance, especially when environmental variables or logistics introduce new stressors. Regulatory uncertainty increases as control strategies become obsolete before they are fully implemented, and critical process parameters may shift unpredictably during technology transfer or scale-up.
To navigate these challenges, organizations are adopting adaptive quality strategies. Predictive quality modeling, using digital twins and machine learning, allows teams to simulate thousands of potential interaction scenarios and forecast failure modes even with incomplete data. Living control systems, powered by AI and continuous process verification, enable dynamic adjustment of specifications and risk priorities as new information emerges. Regulatory agencies are also experimenting with co-evolutionary approaches, such as shared industry databases for risk intelligence and regulatory sandboxes for testing novel quality controls.
Ultimately, compressed innovation cycles demand a fundamental rethinking of quality management. The focus shifts from simply ensuring compliance to actively navigating complexity and anticipating emergent risks. Success in this environment depends on building quality systems that are not only robust and compliant, but also agile and responsive—capable of detecting, understanding, and adapting to surprises as they arise in real time.
Supply Chain Entanglement
Globalization has fundamentally transformed supply chains, creating vast networks that span continents and industries. While this interconnectedness has brought about unprecedented efficiencies and access to resources, it has also introduced a web of hidden interaction vectors—complex, often opaque relationships and dependencies that can amplify both risk and opportunity in ways that are difficult to predict or control.
At the heart of this complexity is the fragmentation of production across multiple jurisdictions. This spatial and organizational dispersion means that disruptions—whether from geopolitical tensions, natural disasters, regulatory changes, or even cyberattacks—can propagate through the network in unexpected ways, sometimes surfacing as quality issues, delays, or compliance failures far from the original source of the problem.
Moreover, the rise of powerful transnational suppliers, sometimes referred to as “Big Suppliers,” has shifted the balance of power within global value chains. These entities do not merely manufacture goods; they orchestrate entire ecosystems of production, labor, and logistics across borders. Their decisions about sourcing, labor practices, and compliance can have ripple effects throughout the supply chain, influencing not just operational outcomes but also the diffusion of norms and standards. This reconsolidation at the supplier level complicates the traditional view that multinational brands are the primary drivers of supply chain governance, revealing instead a more distributed and dynamic landscape of influence.
The hidden interaction vectors created by globalization are further obscured by limited supply chain visibility. Many organizations have a clear understanding of their direct, or Tier 1, suppliers but lack insight into the lower tiers where critical risks often reside. This opacity can mask vulnerabilities such as overreliance on a single region, exposure to forced labor, or susceptibility to regulatory changes in distant markets. As a result, companies may find themselves blindsided by disruptions that originate deep within their supply networks, only becoming apparent when they manifest as operational or reputational crises.
In this environment, traditional risk management approaches are often insufficient. The sheer scale and complexity of global supply chains demand new strategies for mapping connections, monitoring dependencies, and anticipating how shocks in one part of the world might cascade through the system. Advanced analytics, digital tools, and collaborative relationships with suppliers are increasingly essential for uncovering and managing these hidden vectors. Ultimately, globalization has made supply chains more efficient but also more fragile, with hidden interaction points that require constant vigilance and adaptive management to ensure resilience and sustained performance.
Emergence and the Success/Failure Space: Navigating Complexity in System Design
The interplay between emergence and success/failure space reveals a fundamental tension in managing complex systems: our ability to anticipate outcomes is constrained by both the unpredictability of component interactions and the inherent asymmetry between defining success and preventing failure. Emergence is not merely a technical challenge, but a manifestation of how systems oscillate between latent potential and realized risk.
Success space encompasses infinite potential pathways to desired outcomes, characterized by continuous variables like efficiency and adaptability.
Failure space contains discrete, identifiable modes of dysfunction, often easier to consensus-build around than nebulous success metrics.
Emergence complicates this duality. While traditional risk management focuses on cataloging failure modes, emergent behaviors—particularly strong emergence—defy this reductionist approach. Failures can arise not from component breakdowns, but from unexpected couplings between validated subsystems operating within design parameters. This creates a paradox: systems optimized for success space metrics (e.g., throughput, cost efficiency) may inadvertently amplify failure space risks through emergent interactions.
Emergence as a Boundary Phenomenon
Emergent behaviors manifest at the interface of success and failure spaces:
Weak Emergence Predictable through detailed modeling, these behaviors align with traditional failure space analysis. For example, a pharmaceutical plant might anticipate temperature excursion risks in cold chain logistics through FMEA, implementing redundant monitoring systems.
Strong Emergence Unpredictable interactions that bypass conventional risk controls. Consider a validated ERP system that unexpectedly generates phantom batch records when integrated with new MES modules—a failure emerging from software handshake protocols never modeled during individual system validation.
To return to a previous analogy of house purchasing to illustrate this dichotomy: while we can easily identify foundation cracks (failure space), defining the “perfect home” (success space) remains subjective. Similarly, strong emergence represents foundation cracks in system architectures that only become visible after integration.
Reconciling Spaces Through Emergence-Aware Design
To manage this complexity, organizations must:
1. Map Emergence Hotspots Emergence hotspots represent critical junctures where localized interactions generate disproportionate system-wide impacts—whether beneficial innovations or cascading failures. Effectively mapping these zones requires integrating spatial, temporal, and contextual analytics to navigate the interplay between component behaviors and collective outcomes..
2. Implement Ambidextrous Monitoring Combine failure space triggers (e.g., sterility breaches) with success space indicators (e.g., adaptive process capability) – pairing traditional deviation tracking with positive anomaly detection systems that flag beneficial emergent patterns.
3. Cultivate Graceful Success
Graceful success represents a paradigm shift from failure prevention to intelligent adaptation—creating systems that maintain core functionality even when components falter. Rooted in resilience engineering principles, this approach recognizes that perfect system reliability is unattainable, and instead focuses on designing architectures that fail into high-probability success states while preserving safety and quality.
Controlled State Transitions: Systems default to reduced-but-safe operational modes during disruptions.
Decoupled Subsystem Design: Modular architectures prevent cascading failures. This implements the four layers of protection philosophy through physical and procedural isolation.
Dynamic Risk Reconfiguration: Continuously reassess risk priorities using real-time data brings the concept of fail forward into structured learning modes.
This paradigm shift from failure prevention to failure navigation represents the next evolution of quality systems. By designing for graceful success, organizations transform disruptions into structured learning opportunities while maintaining continuous value delivery—a critical capability in an era of compressed innovation cycles and hyperconnected supply chains.
The Emergence Literacy Imperative
This evolution demands rethinking Deming’s “profound knowledge” for the complexity age. Just as failure space analysis provides clearer boundaries, understanding emergence gives us lenses to see how those boundaries shift through system interactions. The organizations thriving in this landscape aren’t those eliminating surprises, but those building architectures where emergence more often reveals novel solutions than catastrophic failures—transforming the success/failure continuum into a discovery engine rather than a risk minefield.
2. Design for Graceful Failure When emergence inevitably occurs, systems should fail into predictable states. For example, you can redesign batch records with:
Modular sections that remain valid if adjacent components fail
Context-aware checklists that adapt requirements based on real-time bioreactor data
Decoupled approvals allowing partial releases while investigating emergent anomalies
3. Harness Beneficial Emergence The most advanced quality systems intentionally foster positive emergence.
The Emergence Imperative
Future-ready quality professionals will balance three tensions:
Prediction AND Adaptation : Investing in simulation while building response agility
Standardization AND Contextualization : Maintaining global standards while allowing local adaptation
Control AND Creativity : Preventing harm while nurturing beneficial emergence
The organizations thriving in this new landscape aren’t those with perfect compliance records, but those that rapidly detect and adapt to emergent patterns. They understand that quality systems aren’t static fortresses, but living networks—constantly evolving, occasionally surprising, and always revealing new paths to excellence.
In this light, Aristotle’s ancient insight becomes a modern quality manifesto: Our systems will always be more than the sum of their parts. The challenge—and opportunity—lies in cultivating the wisdom to guide that “more” toward better outcomes.
The allure of shiny new tools in quality management is undeniable. Like magpies drawn to glittering objects, professionals often collect methodologies and technologies without a cohesive strategy. This “magpie syndrome” creates fragmented systems—FMEA here, 5S there, Six Sigma sprinkled in—that resemble disjointed toolkits rather than coherent ecosystems. The result? Confusion, wasted resources, and quality systems that look robust on paper but crumble under scrutiny. The antidote lies in reimagining quality systems not as static machines but as living organizations that evolve, adapt, and thrive.
The Shift from Machine Logic to Organic Design
Traditional quality systems mirror 20th-century industrial thinking: rigid hierarchies, linear processes, and documents that gather dust. These systems treat organizations as predictable machines, relying on policies to command and procedures to control. Yet living systems—forests, coral reefs, cities—operate differently. They self-organize around shared purpose, adapt through feedback, and balance structure with spontaneity. Deming foresaw this shift. His System of Profound Knowledge—emphasizing psychology, variation, and systems thinking—aligns with principles of living systems: coherence without control, stability with flexibility.
At the heart of this transformation is the recognition that quality emerges not from compliance checklists but from the invisible architecture of relationships, values, and purpose. Consider how a forest ecosystem thrives: trees communicate through fungal networks, species coexist through symbiotic relationships, and resilience comes from diversity, not uniformity. Similarly, effective quality systems depend on interconnected elements working in harmony, guided by a shared “DNA” of purpose.
The Four Pillars of Living Quality Systems
Purpose as Genetic Code Every living system has inherent telos—an aim that guides adaptation. For quality systems, this translates to policies that act as genetic non-negotiables. For pharmaceuticals and medical devices this is “patient safety above all.”. This “DNA” allowed teams to innovate while maintaining adherence to core requirements, much like genes express differently across environments without compromising core traits.
Self-Organization Through Frameworks Complex systems achieve order through frameworks as guiding principles. Coherence emerges from shared intent. Deming’s PDSA cycles and emphasis on psychological safety create similar conditions for self-organization.
Documentation as a Nervous System The enhanced document pyramid—policies, programs, procedures, work instructions, records—acts as an organizational nervous system. Adding a “program” level between policies and procedures bridges the gap between intent and action and can transform static documents into dynamic feedback loops.
Maturity as Evolution Living systems evolve through natural selection. Maturity models serve as evolutionary markers:
Ad-hoc (Primordial): Tools collected like random mutations.
Sustainability: Planning for decade-long impacts, not quarterly audits.
Elegance: Simplifying until it hurts, then relaxing slightly.
Coordination: Cross-pollinating across the organization
Convenience: Making compliance easier than non-compliance.
These principles operationalize Deming’s wisdom. Driving out fear (Point 8) fosters psychological safety, while breaking down barriers (Point 9) enables cross-functional symbiosis.
The Quality Professional’s New Role: Gardener, Not Auditor
Quality professionals must embrace a transformative shift in their roles. Instead of functioning as traditional enforcers or document controllers, we are now called to act as stewards of living systems. This evolution requires a mindset change from one of rigid oversight to one of nurturing growth and adaptability. The modern quality professional takes on new identities such as coach, data ecologist, and systems immunologist—roles that emphasize collaboration, learning, and resilience.
To thrive in this new capacity, practical steps must be taken. First, it is essential to prune toxic practices by eliminating fear-driven reporting mechanisms and redundant tools that stifle innovation and transparency. Quality professionals should focus on fostering trust and streamlining processes to create healthier organizational ecosystems. Next, they must plant feedback loops by embedding continuous learning into daily workflows. For instance, incorporating post-meeting retrospectives can help teams reflect on successes and challenges, ensuring ongoing improvement. Lastly, cross-pollination is key to cultivating diverse perspectives and skills. Rotating staff between quality assurance, operations, and research and development encourages knowledge sharing and breaks down silos, ultimately leading to more integrated and innovative solutions.
By adopting this gardener-like approach, quality professionals can nurture the growth of resilient systems that are better equipped to adapt to change and complexity. This shift not only enhances organizational performance but also fosters a culture of continuous improvement and collaboration.
Thriving, Not Just Surviving
Quality systems that mimic life—not machinery—turn crises into growth opportunities. As Deming noted, “Learning is not compulsory… neither is survival.” By embracing living system principles, we create environments where survival is the floor, and excellence is the emergent reward.
Start small: Audit one process using living system criteria. Replace one control mechanism with a self-organizing principle. Share learnings across your organizational “species.” The future of quality isn’t in thicker binders—it’s in cultivating systems that breathe, adapt, and evolve.