Defining a GMP critical system is an essential aspect of Good Manufacturing Practices (GMP) in the pharmaceutical and medical device industries. A critical system is one that has a direct impact on product quality, safety, and efficacy.
Key Characteristics of GMP Critical Systems
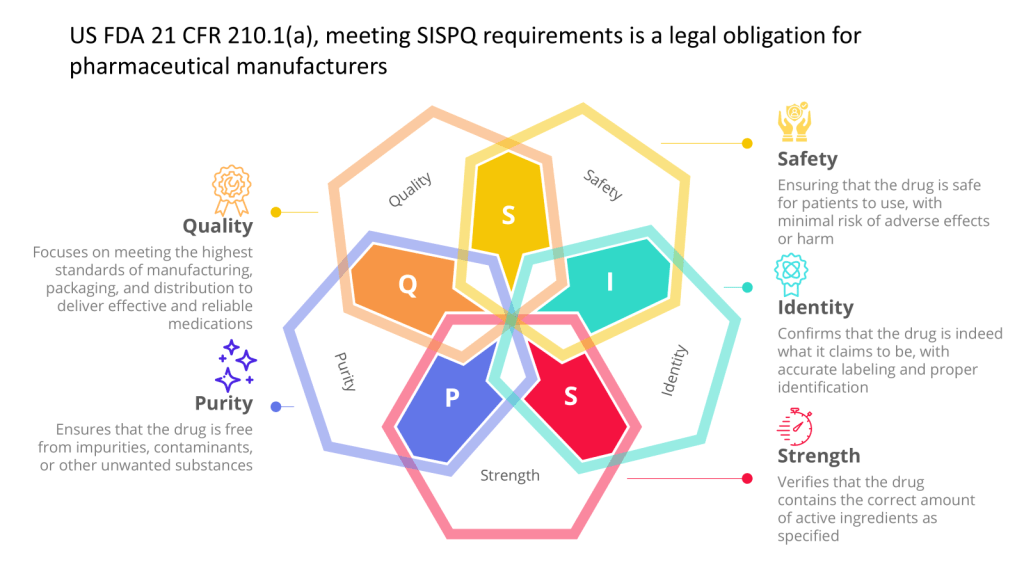
Direct Impact on Product Quality: A critical system is one that can directly affect the quality, safety, or efficacy of the final product.
Influence on Patient Safety: Systems that have a direct or indirect influence on patient safety are considered critical. This is where CPPs come in
Data Integrity: Systems that generate, store, or process data used to determine product SISPQ (e.g. batch quality or are included in batch processing records, stability, data used in a regulatory filing) are critical.
Decision-Making Role: Systems used in the decision process for product release or a regulatory filing are considered critical.
Contact with Products: Equipment or devices that may come into contact with products are often classified as critical.
Continuous Evaluation
It’s important to note that the criticality of systems should be periodically evaluated to ensure they remain in a valid state and compliant with GMP requirements. This includes reviewing the current range of functionality, deviation records, incidents, problems, upgrade history, performance, reliability, security, and validation status reports.
This draft guidance adopts the ICH E3(R1) definitions for protocol deviation and important protocol deviation, providing more standardized terminology, which is a great thing. Avoiding the term “protocol violation”, it primarily uses “protocol deviation” and “important protocol deviation.”
The FDA guidance provides detailed sections on the roles and responsibilities of investigators, sponsors, and IRBs in monitoring, mitigating, and reporting protocol deviations. It as specific recommendations for reporting protocol deviations to sponsors, IRBs, and FDA, including timelines and methods.
It mostly seems a good application of a quality-by-design approach, focusing on critical-to-quality factors and risk-based monitoring for clinical studies. Hopefully it will help clear up confusion in this area.
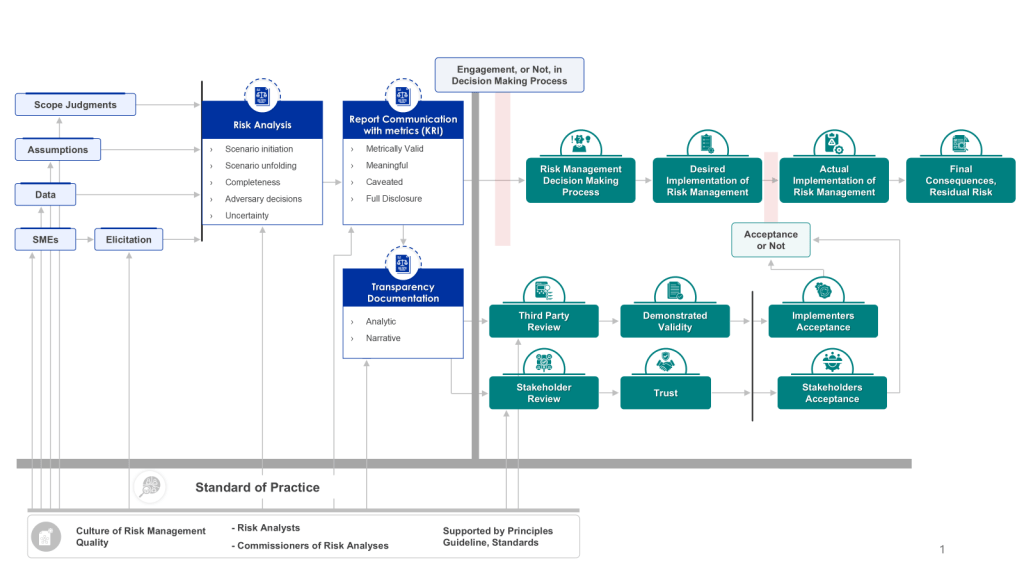
Effective risk analysis is crucial for informed decision-making and robust risk management. Simply conducting a risk analysis is not enough; its effectiveness in engaging the risk management decision-making process is paramount. This effectiveness is largely driven by the transparency and documentation of the analysis, which supports both stakeholder and third-party reviews. Let’s explore how we can measure this effectiveness and why it matters.
The Importance of Transparency and Documentation
Transparency and documentation form the backbone of an effective risk analysis process. They ensure that the methodology, assumptions, and results of the analysis are clear and accessible to all relevant parties. This clarity is essential for:
To improve the effectiveness of risk analysis through better transparency and documentation:
Standardize Risk Reporting
Develop standardized templates and formats for risk reports to ensure consistency and completeness. This standardization facilitates easier comparison and analysis across different time periods or business units.
Implement a Risk Taxonomy
Create a common language for risk across the organization. A well-defined risk taxonomy ensures that all stakeholders understand and interpret risk information consistently.
Leverage Visualization Tools
Utilize data visualization techniques to present risk information in an easily digestible format. Visual representations can make complex risk data more accessible to a broader audience, enhancing engagement in the decision-making process.
Maintain a Comprehensive Audit Trail
Document all steps of the risk analysis process, including data sources, methodologies, assumptions, and decision rationales. This audit trail is crucial for both internal reviews and external audits.
Foster a Culture of Transparency
Encourage open communication about risks throughout the organization. This cultural shift can lead to more honest and accurate risk reporting, ultimately improving the quality of risk analysis.
Conclusion
Measuring the effectiveness of risk analysis in engaging the risk management decision-making process is crucial for organizations seeking to optimize their risk management strategies. By focusing on transparency and documentation, and implementing key metrics to track performance, organizations can ensure that their risk analysis efforts truly drive informed decision-making and robust risk management.
Remember, the goal is not just to conduct risk analysis, but to make it an integral part of the organization’s decision-making fabric. By continuously measuring and improving the effectiveness of risk analysis, organizations can build resilience, enhance stakeholder trust, and navigate uncertainties with greater confidence.
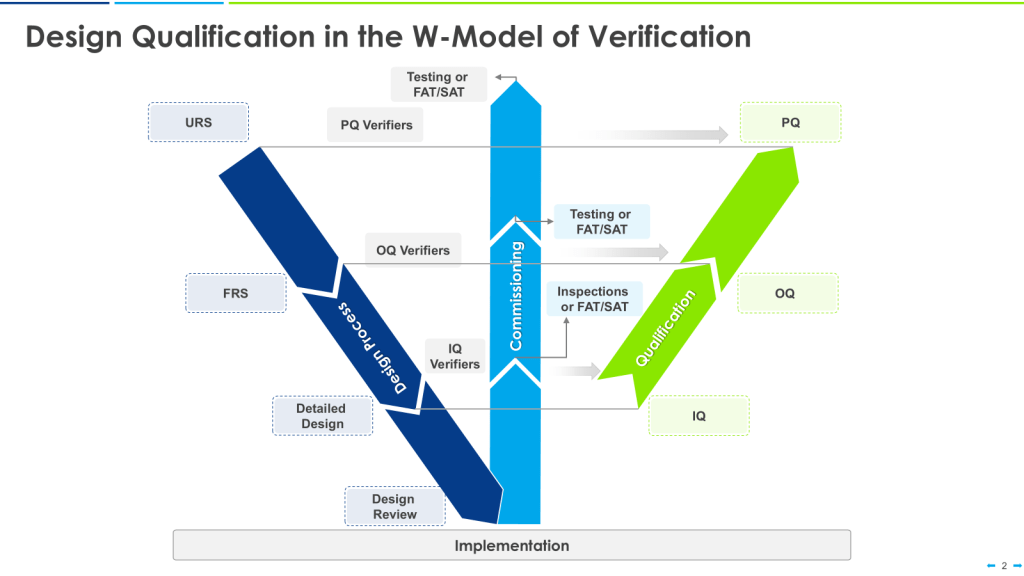
A critical step in ensuring the quality and safety of processes as part of verification is Design Review, which is sometimes expanded to Design Qualification.
Design Review is a systematic, documented examination of a proposed design to evaluate its adequacy and identify potential issues early in the development process. Here’s how to conduct an effective Design Review:
Plan Systematically: Schedule reviews at appropriate stages of development, ensuring they align with your project timeline.
Involve the Right People: Include representatives from all relevant functions and an independent reviewer not directly responsible for the design stage being evaluated.
Focus on Critical Aspects: Prioritize design elements that directly impact product quality and patient safety.
Document Thoroughly: Record all findings, including the design under review, participants, date, and any proposed actions.
Iterate as Needed: Conduct reviews iteratively as supplier design documents are published, allowing for early issue identification and correction.
Design Qualification: Verifying Suitability
Design Qualification (DQ) is the documented verification that the proposed design of facilities, equipment, or systems is suitable for its intended purpose. Here’s how to implement DQ effectively:
Develop User Requirements: Create a detailed User Requirements Specification (URS) outlining what the equipment or system is expected to do.
Create Functional Specifications: Translate user requirements into technical specifications that guide the design process.
Perform Risk Assessment: Identify potential risks associated with the design and develop mitigation strategies.
Review Design Specifications: Ensure the design meets all specified requirements, including GMP and regulatory standards.
Document and Approve: Formally document the DQ process and obtain approval from key stakeholders, including quality assurance personnel.
Integrating Design Review and DQ
To maximize the effectiveness of these processes:
Use a Risk-Based Approach: Prioritize efforts based on the level of risk to product quality and patient safety.
Leverage Subject Matter Experts: Involve SMEs from the start to contribute their expertise throughout the process.
Implement Change Management: Establish a robust system to manage design changes effectively and avoid late-stage issues.
Ensure Quality Oversight: Have Quality Assurance provide oversight to maintain compliance with current regulations and GMP requirements.
Document Comprehensively: Maintain thorough records of all reviews, qualifications, and decisions made during the process.
Implementing a systematic approach to Design Review and Design Qualification not only helps meet regulatory expectations but also contributes to operational efficiency and product excellence. As the pharmaceutical landscape evolves, staying committed to these foundational practices will remain crucial for success in this highly regulated industry.
User requirements are typically divided into several categories to ensure comprehensive coverage of all aspects of product development, manufacturing, and quality control and to help guide the risk-based approach to verification.
Quality requirements focus on ensuring that the product meets all necessary quality standards and regulatory compliance. This category includes:
Good Manufacturing Practices (GMP) compliance, including around cleaning, cross-contamination, etc to ensure compliance with various regulations such as FDA guidelines, EU GMP, and ICH standards.
Documentation and record-keeping standards
Contamination control strategies are a key part of quality requirements, as they are essential for maintaining product quality and patient safety.
Data integrity requirements fall under this category, as they are crucial for ensuring the quality and reliability of data.
Not everyone advocates for this breakdown but I am a huge proponent as it divides the product specific requirements for the more standard must’s of meeting the cGMPs that are not product specific. This really helps when you are a multi-product facility and it helps define what is in the PQ versus what is in the PPQ.
Safety User Requirements
Safety requirements address the safety of personnel, patients, and the environment. They encompass:
Occupational health and safety measures
Environmental protection protocols
Patient safety considerations in product design
General User Requirements
General requirements cover broader aspects of the manufacturing system and facility. These may include:
Facility design and layout
Equipment specifications
Utility requirements (e.g., power, water, HVAC)
Maintenance procedures
By categorizing user requirements in this way, pharmaceutical companies can ensure a comprehensive approach to product development and manufacturing, addressing all critical aspects from product quality to regulatory compliance and safety. This will help drive appropriate verification.