Let us turn our failure space model, and level of problems, to deviations in a clinical trial. This is one of those areas that regulations and tribal practice have complicated, perhaps needlessly. It is also complicated by the different players of clinical sites, sponsor, and usually these days a number of Contract Research Organizations (CRO).
What is a Protocol Deviation?
Protocol deviation is any change, divergence, or departure from the study design or procedures defined in the approved protocol.
Protocol deviations may include unplanned instances of protocol noncompliance. For example, situations in which the clinical investigator failed to perform tests or examinations as required by the protocol or failures on the part of subjects to complete scheduled visits as required by the protocol, would be considered protocol deviations.
In the case of deviations which are planned exceptions to the protocol such deviations should be reviewed and approved by the IRB, the sponsor, and by the FDA for medical devices, prior to implementation, unless the change is necessary to eliminate apparent immediate hazards to the human subjects (21 CFR 312.66), or to protect the life or physical well-being of the subject (21 CFR 812.150(a)(4)).
The FDA, July 2020. Compliance Program Guidance Manual for Clinical Investigator Inspections (7348.811).
In assessing protocol deviations/violations, the FDA instructs field staff to determine whether changes to the protocol were: (1) documented by an amendment, dated, and maintained with the protocol; (2) reported to the sponsor (when initiated by the clinical investigator); and (3) approved by the IRB and FDA (if applicable) before implementation (except when necessary to eliminate apparent immediate hazard(s) to human subjects).
| Regulation/Guidance | States |
| ICH E-6 (R2) Section 4.5.1-4.5.4 | 4.5.1“trial should be conducted in compliance with the protocol agreed to by the sponsor and, if required by the regulatory authorities…” 4.5.2 The investigator should not implement any deviation from, or changes of, the protocol without agreement by the sponsor and prior review and documented approval/favorable opinion from the IRB/IEC of an amendment, except where necessary to eliminate an immediate hazard(s) to trial subjects, or when the change(s) involves only logistical or administrative aspects of the trial (e.g., change in monitor(s), change of telephone number(s)). 4.5.3 The investigator, or person designated by the investigator, should document and explain any deviation from the approved protocol. 4.5.4 The investigator may implement a deviation from, or a change in, the protocol to eliminate an immediate hazard(s) to trial subjects without prior IRB/IEC approval/favorable opinion. |
| ICH E3, section 9.6 | The sponsor should describe the quality management approach implemented in the trial and summarize important deviations from the predefined quality tolerance limits and remedial actions taken in the clinical study report |
| 21CFR 312.53(vi) (a) | investigators selected “Will conduct the study(ies) in accordance with the relevant, current protocol(s) and will only make changes in a protocol after notifying the sponsor, except when necessary to protect the safety, the rights, or welfare of subjects.” |
| 21CFR 56.108(a) | IRB shall….ensur[e] that changes in approved research….may not be initiated without IRB review and approval except where necessary to eliminate apparent immediate hazards to the human subjects. |
| 21 CFR 56.108(b) | “IRB shall….follow written procedures for ensuring prompt reporting to the IRB, appropriate institutional officials, and the Food and Drug Administration of… any unanticipated problems involving risks to human subjects or others…[or] any instance of serious or continuing noncompliance with these regulations or the requirements or determinations of the IRB.” |
| 45 CFR 46.103(b)(5) | Assurances applicable to federally supported or conducted research shall at a minimum include….written procedures for ensuring prompt reporting to the IRB….[of] any unanticipated problems involving risks to subjects or others or any serious or continuing noncompliance with this policy or the requirements or determinations of the IRB. |
| FDA Form-1572 (Section 9) | lists the commitments the investigator is undertaking in signing the 1572 wherein the clinical investigator agrees “to conduct the study(ies) in accordance with the relevant, current protocol(s) and will only make changes in a protocol after notifying the sponsor, except when necessary to protect the safety, the rights, or welfare of subjects… [and] not to make any changes in the research without IRB approval, except where necessary to eliminate apparent immediate hazards to the human subjects.” |
How Protocol Deviations are Implemented
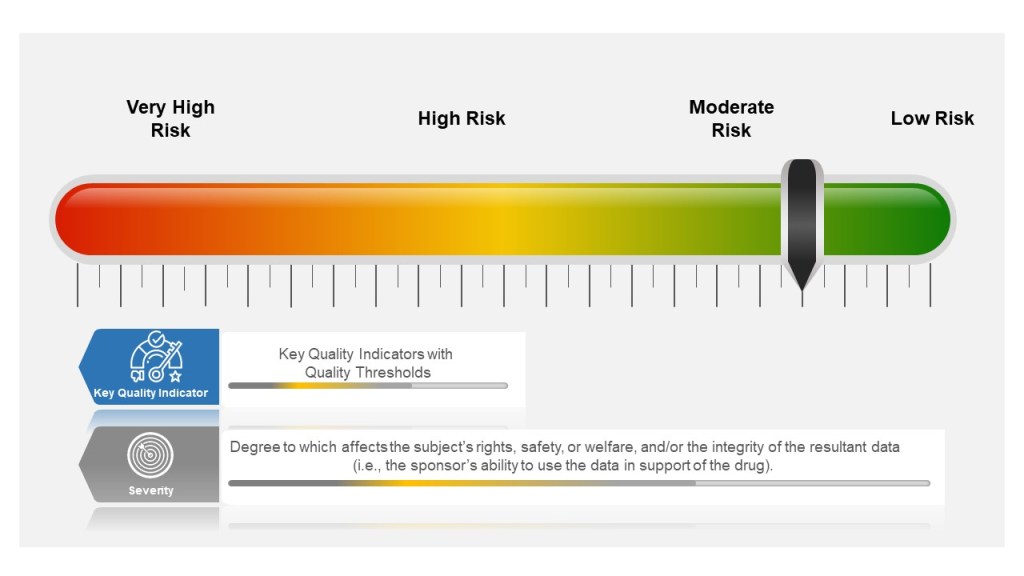
Many companies tend to have a failure scale built into their process, differentiating between protocol deviations and violations based on severity. Others use a minor, major, and even critical scale to denote differences in severity. The axis here for severity is the degree to which affects the subject’s rights, safety, or welfare, and/or the integrity of the resultant data (i.e., the sponsor’s ability to use the data in support of the drug).
Other companies divide into protocol deviations and violations:
- Protocol Deviation: A protocol deviation occurs when, without significant consequences, the activities on a study diverge from the IRB-approved protocol, e.g., missing a visit window because the subject is traveling. Not as serious as a protocol violation.
- Protocol Violation: A divergence from the protocol that materially (a) reduces the quality or completeness of the data, (b) makes the ICF inaccurate, or (c) impacts a subject’s safety, rights or welfare. Examples of protocol violations may include: inadequate or delinquent informed consent; inclusion/exclusion criteria not met; unreported SAEs; improper breaking of the blind; use of prohibited medication; incorrect or missing tests; mishandled samples; multiple visits missed or outside permissible windows; materially inadequate record-keeping; intentional deviation from protocol, GCP or regulations by study personnel; and subject repeated noncompliance with study requirements.
This is probably a place when nomenclature can serve to get in the way, rather than provide benefit. The EMA says pretty much the same in “ICH guideline E3 – questions and answers (R1).“
Principles of Events in Clinical Practice
- Severity of the event is based on degree to which affects the subject’s rights, safety, or welfare, and/or the integrity of the resultant data
- Events (problems, deviations, etc) will happen at all levels of a clinical practice (Sponsor, CRO, Site, etc)
- Events happen beyond the Protocol. These need to be managed appropriately as well.
- The event needs to be categorized, evaluated and trended by the sponsor
Severity of the Event
Starting in the study planning stage, ICH E6(R2) GCP requires sponsors to identify risks to critical study processes and study data and to evaluate these risks based on likelihood, detectability and impact on subject safety and data integrity.
Sponsors then establish key quality indicators (KQIs) and quality tolerance thresholds. KQI is really just a key risk indicator and should be treated similarly.
Study events that exceed the risk threshold should trigger an evaluation to determine if action is needed. In this way, sponsors can proactively manage risk and address protocol noncompliance.
The best practice here is to have a living risk assessment for each study. Evaluate across studies to understand your overall organization risk, and look for opportunities for wide-scale mitigations. Feedup into your risk register.
Where the Event happens
Deviations in the clinical space are a great example of the management of supplier events, and at the end of the day there is little difference between a GMP supplier event management, a GLP or a GCP. The individual requirements might be different but the principles and the process are the same.
Each entity in the trial organization should have their own deviation system where they investigate deviations, performing root cause investigation and enacting CAPAs.
This is where it starts to get tricky. first of all, not all sites have the infrastructure to do this well. Second the nature of reporting, usually through the Electronic Data Capture (EDC) system, can lead to balkanization at the site. Site’s need to have strong compliance programs through compiling deviation details into a single sitewide system that allows the site to trend deviations across studies in addition to following sponsor reporting requirements.
Unfortunately too many site’s rely on the sponsor’s program. Sponsors need to be evaluating the strength of this program during site selection and through auditing.
Events Happen
Deviations should be to all process, procedure and plans, and just not the protocol.
Categorization and Trending
Categorizing deviations is usually a pain point and an area where more consistency needs to be driven. I recommend first having a good standard set of categorizations. The industry would benefit from adopting a standard, and I think Norman Goldfarb’s proposal is still the best.
Once you have categories, and understand to your KQIs and other aspects you need to make sure they are consistently done. The key mechanisms of this are:
- Training
- Monitoring (in all its funny permutations)
- Periodic evaluations and Trending
Deviations should be trended, at a minimum, in several ways:
- Per site per study
- Per site all activities
- All sites per study
- All sites all activities
And remember, trending doesn’t count of you do not analyze the problem and take appropriate CAPAs.
This will allow trends to be identified and appropriate corrective and preventive actions identified to systematically improve.

3 thoughts on “The Failure Space of Clinical Trials – Protocol Deviations and Events”