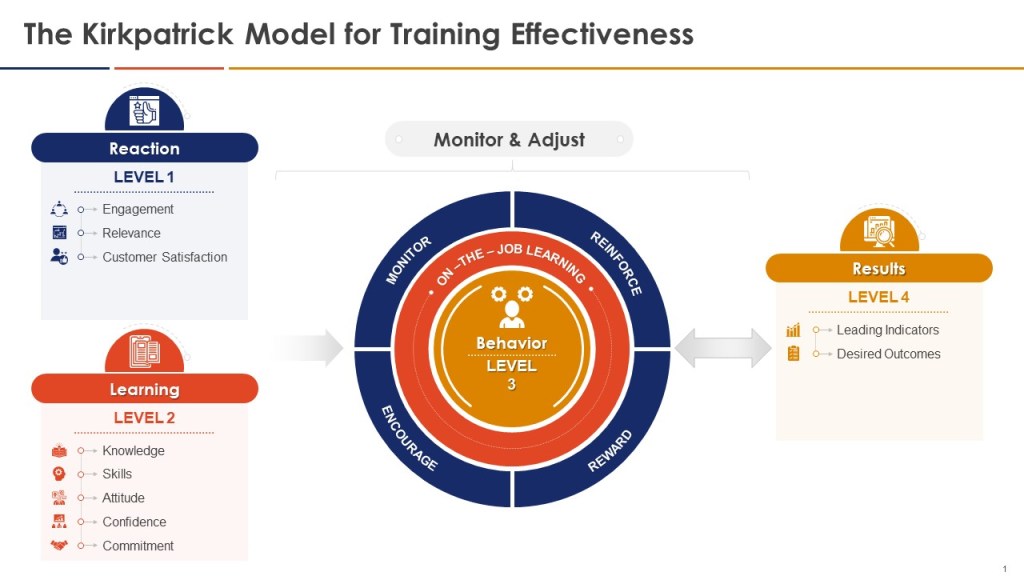
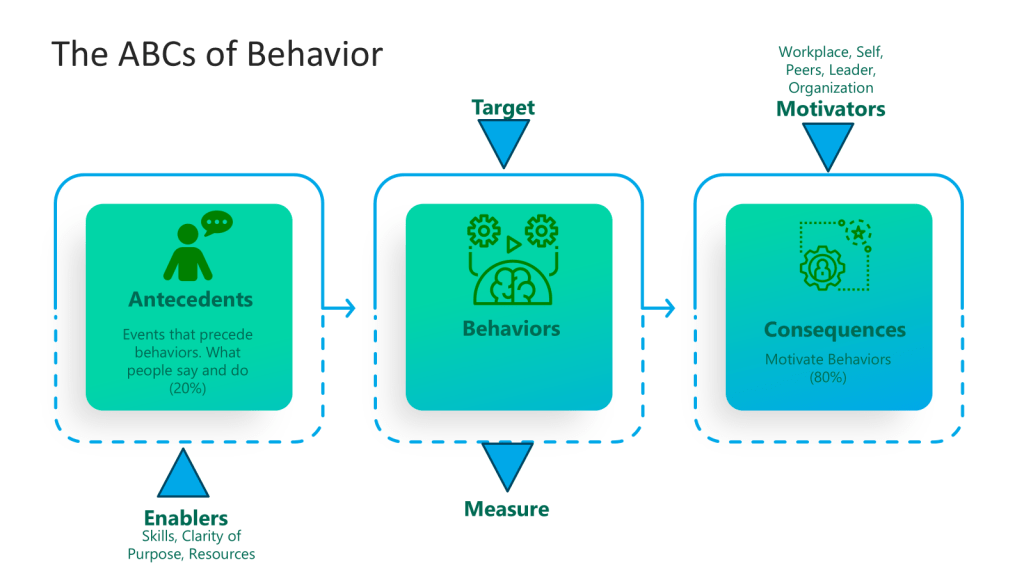
When thinking about the training program you can add the Kilpatrick model to the mix and build from there. This allows a view across the training system to drive for an effective training program.
GMP Training Metrics Framework Aligned with Kirkpatrick’s Model
Kirkpatrick Level
Category
Metric Type
Example
Purpose
Data Source
Regulatory Alignment
Level 1: Reaction
KPI
Leading
% Training Satisfaction Surveys Completed
Measures engagement and perceived relevance of GMP training
LMS (Learning Management System)
ICH Q10 Section 2.7 (Training Effectiveness)
KRI
Leading
% Surveys with Negative Feedback (<70%)
Identifies risk of disengagement or poor training design
Survey Tools
FDA Quality Metrics Reporting (2025 Draft)
KBI
Leading
Participation in Post-Training Feedback
Encourages proactive communication about training gaps
Attendance Logs
EU GMP Chapter 2 (Personnel Training)
Level 2: Learning
KPI
Leading
Pre/Post-Training Quiz Pass Rate (≥90%)
Validates knowledge retention of GMP principles
Assessment Software
21 CFR 211.25 (Training Requirements)
KRI
Leading
% Trainees Requiring Remediation (>15%)
Predicts future compliance risks due to knowledge gaps
LMS Remediation Reports
FDA Warning Letters (Training Deficiencies)
KBI
Lagging
Reduction in Knowledge Assessment Retakes
Validates long-term retention of GMP concepts
Training Records
ICH Q7 Section 2.12 (Training Documentation)
Level 3: Behavior
KPI
Leading
Observed GMP Compliance Rate During Audits
Measures real-time application of training in daily workflows
Audit Checklists
FDA 21 CFR 211 (cGMP Compliance)
KRI
Leading
Near-Miss Reports Linked to Training Gaps
Identifies emerging behavioral risks before incidents occur
QMS (Quality Management System)
ISO 9001:2015 Clause 10.2 (Nonconformity)
KBI
Leading
Frequency of Peer-to-Peer Knowledge Sharing
Encourages a culture of continuous learning and collaboration
Meeting Logs
ICH Q10 Section 3.2.3 (Knowledge Management)
Level 4: Results
KPI
Lagging
% Reduction in Repeat Deviations Post-Training
Quantifies training’s impact on operational quality
Deviation Management Systems
FDA Quality Metrics (Batch Rejection Rate)
KRI
Lagging
Audit Findings Related to Training Effectiveness
Reflects systemic training failures impacting compliance
Regulatory Audit Reports
EU GMP Annex 15 (Qualification & Validation)
KBI
Lagging
Employee Turnover
Assesses cultural impact of training on staff retention
HR Records
ICH Q10 Section 1.5 (Management Responsibility)
Kirkpatrick Model Integration
Level 1 (Reaction):
Leading KPI: Track survey completion to ensure trainees perceive value in GMP content.
Leading KRI: Flag facilities with >30% negative feedback for immediate remediation .
Level 2 (Learning):
Leading KPI: Require ≥90% quiz pass rates for high-risk roles (e.g., aseptic operators) .
Lagging KBI: Retake rates >20% trigger refresher courses under EU GMP Chapter 3 .
Level 3 (Behavior):
Leading KPI: <95% compliance during audits mandates retraining per 21 CFR 211.25 .
Leading KRI: >5 near-misses/month linked to training gaps violates FDA’s “state of control” .
Level 4 (Results):
Lagging KPI: <10% reduction in deviations triggers CAPA under ICH Q10 Section 4.3 .
FDA Quality Metrics: Level 4 KPIs (e.g., deviation reduction) align with FDA’s 2025 focus on “sustainable compliance” .
ICH Q10: Level 3 KBIs (peer knowledge sharing) support “continual improvement of process performance” .
EU GMP: Level 2 KRIs (remediation rates) enforce Annex 11’s electronic training documentation requirements .
By integrating Kirkpatrick’s levels with GMP training metrics, organizations bridge knowledge acquisition to measurable quality outcomes while meeting global regulatory expectations.
Predicts compliance with FDA 21 CFR 211.100 (process control)
FDA 21 CFR 211, ICH Q10, ICH Q9
Lagging
Average Time to Close Change Requests
Validates efficiency of change implementation (EudraLex Annex 15)
EU GMP Annex 15
KRI
Leading
Unresolved CAPAs Linked to Change Requests
Identifies systemic risks before deviations occur (FDA Warning Letters)
21 CFR 211.22, ICH Q7
Lagging
Repeat Deviations Post-Change
Reflects failure to address root causes (FDA 483 Observations)
21 CFR 211.192
KBI
Leading
Cross-Functional Review Participation Rate
Encourages proactive collaboration in change evaluation
ICH Q10 Section 3.2.3
Lagging
Reduction in Documentation Errors Post-Training
Validates effectiveness of staff competency programs
EU 1252/2014 Article 14
Key Performance Indicators (KPIs)
Leading KPI:
Change Requests with Completed Risk Assessments: Measures proactive compliance with FDA requirements for risk-based change evaluation. A rate <90% triggers quality reviews.
Lagging KPI:
Time to Close Changes: Benchmarks against EMA’s 30-day resolution expectation for critical changes. Prolonged closure (>45 days) indicates process bottlenecks.
Key Risk Indicators (KRIs)
Leading KRI:
Unresolved CAPAs: Predicts validation gaps; >5 open CAPAs per change violates FDA’s “state of control” mandate.
Lagging KRI:
Repeat Deviations: >3 repeat deviations quarterly triggers mandatory revalidation per FDA 21 CFR 211.180.
Documentation Errors: Post-training error reduction <30% prompts requalification under EU GMP Chapter 4.
Implementation Guidance
Align with Regulatory Thresholds: Set leading KPI targets using FDA’s 2025 draft guidance: ≥95% risk assessment completion for high-impact changes.
Automate Tracking: Integrate metrics with eQMS software to monitor CAPA aging (leading KRI) and deviation trends (lagging KRI) in real time.
Link to Training: Tie lagging KBIs to annual GMP refresher courses, as required by EU 1252/2014 Article 14.
Why It Matters: Leading metrics enable proactive mitigation of change-related risks (e.g., unresolved CAPAs predicting audit failures), while lagging metrics validate adherence to FDA’s lifecycle approach for process validation. Balancing both ensures compliance with 21 CFR 211’s “state of control” mandate while fostering continuous improvement.
Understanding how to measure success and risk is critical for organizations aiming to achieve strategic objectives. As we develop Quality Plans and Metric Plans it is important to explore the nuances of leading and lagging metrics, define Key Performance Indicators (KPIs), Key Behavioral Indicators (KBIs), and Key Risk Indicators (KRIs), and explains how these concepts intersect with Objectives and Key Results (OKRs).
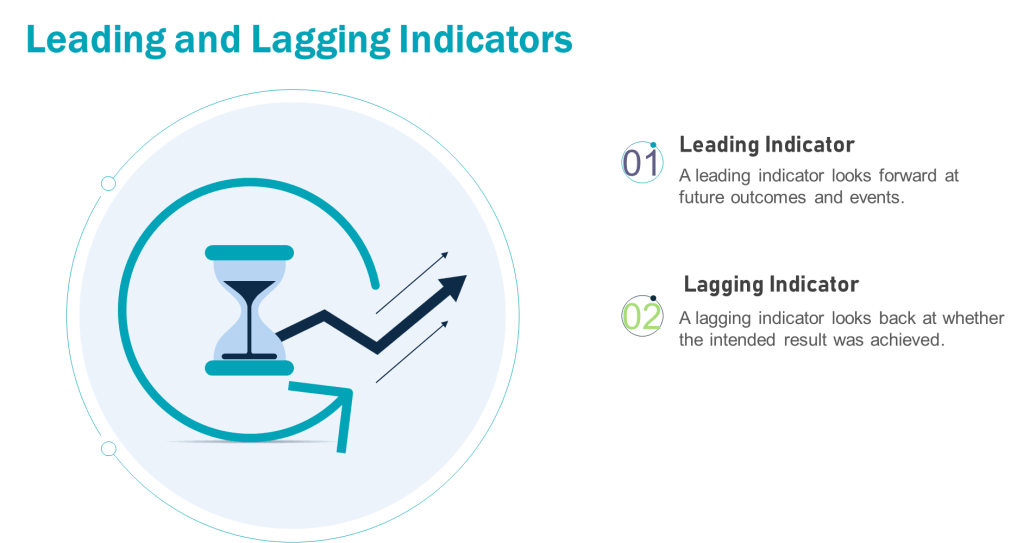
Leading vs. Lagging Metrics: A Foundation
Leading metrics predict future outcomes by measuring activities that drive results. They are proactive, forward-looking, and enable real-time adjustments. For example, tracking employee training completion rates (leading) can predict fewer operational errors.
Lagging metrics reflect historical performance, confirming whether quality objectives were achieved. They are reactive and often tied to outcomes like batch rejection rates or the number of product recalls. For example, in a pharmaceutical quality system, lagging metrics might include the annual number of regulatory observations, the percentage of batches released on time, or the rate of customer complaints related to product quality. These metrics provide a retrospective view of the quality system’s effectiveness, allowing organizations to assess their performance against predetermined quality goals and industry standards. They offer limited opportunities for mid-course corrections
The interplay between leading and lagging metrics ensures organizations balance anticipation of future performance with accountability for past results.
Defining KPIs, KRIs, and KBIs
Key Performance Indicators (KPIs)
KPIs measure progress toward Quality System goals. They are outcome-focused and often tied to strategic objectives.
Leading KPI Example: Process Capability Index (Cpk) – This measures how well a process can produce output within specification limits. A higher Cpk could indicate fewer products requiring disposition.
Lagging KPI Example: Cost of Poor Quality (COPQ) -The total cost associated with products that don’t meet quality standards, including testing and disposition cost.
Key Risk Indicators (KRIs)
KRIs monitor risks that could derail objectives. They act as early warning systems for potential threats. Leading KRIs should trigger risk assessments and/or pre-defined corrections when thresholds are breached.
Leading KRI Example: Unresolved CAPAs (Corrective and Preventive Actions) – Tracks open corrective actions for past deviations. A rising number signals unresolved systemic issues that could lead to recurrence
Lagging KRI Example: Repeat Deviation Frequency – Tracks recurring deviations of the same type. Highlights ineffective CAPAs or systemic weaknesses
Key Behavioral Indicators (KBIs)
KBIs track employee actions and cultural alignment. They link behaviors to Quality System outcomes.
Leading KBI Example: Frequency of safety protocol adherence (predicts fewer workplace accidents).
Lagging KBI Example: Employee turnover rate (reflects past cultural challenges).
Applying Leading and Lagging Metrics to KPIs, KRIs, and KBIs
Each metric type can be mapped to leading or lagging dimensions:
KPIs: Leading KPIs drive action while lagging KPIs validate results
KRIs: Leading KRIs identify emerging risks while lagging KRIs analyze past incidents
KBIs: Leading KBIs encourage desired behaviors while lagging KBIs assess outcomes
Proactively ensures continued process verification aligns with validation master plans
Validation tracking systems
Lagging
Annual audit findings related to validation drift
Confirms adherence to regulator’s “state of control” requirements
Internal/regulatory audit reports
KRI
Leading
Open CAPAs linked to FUSe(P) validation gaps
Identifies unresolved systemic risks affecting process robustness
Quality management systems (QMS)
Lagging
Repeat deviations in validated batches
Reflects failure to address root causes post-validation
Deviation management systems
KBI
Leading
Cross-functional review of process monitoring trends
Encourages proactive behavior to maintain validation state
Meeting minutes, action logs
Lagging
Reduction in human errors during requalification
Validates effectiveness of training/behavioral controls
Training records, deviation reports
This framework operationalizes a focus on data-driven, science-based programs while closing gaps cited in recent Warning Letters.
Goals vs. OKRs: Alignment with Metrics
Goals are broad, aspirational targets (e.g., “Improve product quality”). OKRs (Objectives and Key Results) break goals into actionable, measurable components:
Objective: Reduce manufacturing defects.
Key Results:
Decrease batch rejection rate from 5% to 2% (lagging KPI).
Train 100% of production staff on updated protocols by Q2 (leading KPI).
Reduce repeat deviations by 30% (lagging KRI).
KPIs, KRIs, and KBIs operationalize OKRs by quantifying progress and risks. For instance, a leading KRI like “number of open CAPAs” (Corrective and Preventive Actions) informs whether the OKR to reduce defects is on track.
More Pharmaceutical Quality System Examples
Leading Metrics
KPI: Percentage of staff completing GMP training (predicts adherence to quality standards).
KRI: Number of unresolved deviations in the CAPA system (predicts compliance risks).
KBI: Daily equipment calibration checks (predicts fewer production errors).
Lagging Metrics
KPI: Batch rejection rate due to contamination (confirms quality failures).
KRI: Regulatory audit findings (reflects past non-compliance).
KBI: Employee turnover in quality assurance roles (indicates cultural or procedural issues).
Metric Type
Purpose
Leading Example
Lagging Example
KPI
Measure performance outcomes
Training completion rate
Quarterly profit margin
KRI
Monitor risks
Open CAPAs
Regulatory violations
KBI
Track employee behaviors
Safety protocol adherence frequency
Employee turnover rate
Building Effective Metrics
Align with Strategy: Ensure metrics tie to Quality System goals. For OKRs, select KPIs/KRIs that directly map to key results.
Balance Leading and Lagging: Use leading indicators to drive proactive adjustments and lagging indicators to validate outcomes.
Pharmaceutical Focus: In quality systems, prioritize metrics like right-first-time rate (leading KPI) and repeat deviation rate (lagging KRI) to balance prevention and accountability.
By integrating KPIs, KRIs, and KBIs into OKRs, organizations create a feedback loop that connects daily actions to long-term success while mitigating risks. This approach transforms abstract goals into measurable, actionable pathways—a critical advantage in regulated industries like pharmaceuticals.
Understanding these distinctions empowers teams to not only track performance but also shape it proactively, ensuring alignment with both immediate priorities and strategic vision.
Risk-based thinking is a crucial component of modern quality management systems and consists of four key aspects: anticipate, monitor, respond, and learn. Each aspect ensures an organization can effectively manage and mitigate risks, enhancing overall performance and reliability.
Anticipate
Anticipating risks involves proactively identifying and analyzing potential risks that could impact the organization’s operations or objectives. This step is about foreseeing problems before they occur and planning how to address them. It requires a thorough understanding of the organization’s processes, the external and internal factors that could affect these processes, and the potential consequences of various risks. By anticipating risks, organizations can prepare more effectively and prevent many issues from occurring.
Monitor
Monitoring involves continuously observing and tracking the operational environment to detect risk indicators early. This ongoing process helps catch deviations from expected outcomes or standards, which could indicate the emergence of a risk. Effective monitoring relies on establishing metrics that help to quickly and accurately identify when things are starting to veer off course. This real-time data collection is crucial for enabling timely responses to potential threats.
Respond
Responding to risks is about taking appropriate actions to manage or mitigate identified risks based on their severity and potential impact. This step involves implementing the planned risk responses that were developed during the anticipation phase. The effectiveness of these responses often depends on the speed and decisiveness of the actions taken. Responses can include adjusting processes, reallocating resources, or activating contingency plans. The goal is to minimize the organization’s and its stakeholders’ negative impact.
Learn
Learning from the management of risks is a critical component that closes the loop of risk-based thinking. This aspect involves analyzing the outcomes of risk responses and understanding what worked well and what did not. Learning from these experiences is essential for continuous improvement. It helps organizations refine risk management processes, improve response strategies, and better prepare for future risks. This iterative learning process ensures that risk management efforts are increasingly effective over time.
The four aspects of risk-based thinking—anticipate, monitor, respond, and learn—form a continuous cycle that helps organizations manage uncertainties proactively. This approach protects the organization from potential downsides and enables it to seize opportunities that arise from a well-understood risk landscape. Organizations can enhance their resilience and adaptability by embedding these practices into everyday operations.
Implementing Risk-Based Thinking
1. Understand the Concept of Risk-Based Thinking
Risk-based thinking involves a proactive approach to identifying, analyzing, and addressing risks. This mindset should be ingrained in the organization’s culture and used as a basis for decision-making.
2. Identify Risks and Opportunities
Identify potential risks and opportunities. This can be achieved through various methods such as SWOT analysis, brainstorming sessions, and process mapping. It’s crucial to involve people at all levels of the organization since they can provide diverse perspectives on potential risks and opportunities.
3. Analyze and Prioritize Risks
Once risks and opportunities are identified, they should be analyzed to understand their potential impact and likelihood. This analysis will help prioritize which risks need immediate attention and which opportunities should be pursued.
4. Plan and Implement Responses
After prioritizing, develop strategies to address these risks and opportunities. Plans should include preventive measures for risks and proactive steps to seize opportunities. Integrating these plans into the organization’s overall strategy and daily operations is important to ensure they are effective.
5. Monitor and Review
Implementing risk-based thinking is not a one-time activity but an ongoing process. Regular monitoring and reviewing of risks, opportunities, and the effectiveness of responses are crucial. This can be done through regular audits, performance evaluations, and feedback mechanisms. Adjustments should be made based on these reviews to improve the risk management process.
6. Learn and Improve
Organizations should learn from their experiences in managing risks and opportunities. This involves analyzing what worked well and what didn’t and using this information to improve future risk management efforts. Continuous improvement should be a key goal, aligning with the Plan-Do-Check-Act (PDCA) cycle.
Training and cultural adaptation are necessary to implement risk-based thinking effectively. All employees should be trained on the principles of risk-based thinking and how to apply them in their roles. Creating a culture encouraging open communication about risks and supporting risk-taking within defined limits is also vital.
Let us turn our failure space model, and level of problems, to deviations in a clinical trial. This is one of those areas that regulations and tribal practice have complicated, perhaps needlessly. It is also complicated by the different players of clinical sites, sponsor, and usually these days a number of Contract Research Organizations (CRO).
What is a Protocol Deviation?
Protocol deviation is any change, divergence, or departure from the study design or procedures defined in the approved protocol.
Protocol deviations may include unplanned instances of protocol noncompliance. For example, situations in which the clinical investigator failed to perform tests or examinations as required by the protocol or failures on the part of subjects to complete scheduled visits as required by the protocol, would be considered protocol deviations.
In the case of deviations which are planned exceptions to the protocol such deviations should be reviewed and approved by the IRB, the sponsor, and by the FDA for medical devices, prior to implementation, unless the change is necessary to eliminate apparent immediate hazards to the human subjects (21 CFR 312.66), or to protect the life or physical well-being of the subject (21 CFR 812.150(a)(4)).
The FDA, July 2020. Compliance Program Guidance Manual for Clinical Investigator Inspections (7348.811).
In assessing protocol deviations/violations, the FDA instructs field staff to determine whether changes to the protocol were: (1) documented by an amendment, dated, and maintained with the protocol; (2) reported to the sponsor (when initiated by the clinical investigator); and (3) approved by the IRB and FDA (if applicable) before implementation (except when necessary to eliminate apparent immediate hazard(s) to human subjects).
Regulation/Guidance
States
ICH E-6 (R2) Section 4.5.1-4.5.4
4.5.1“trial should be conducted in compliance with the protocol agreed to by the sponsor and, if required by the regulatory authorities…” 4.5.2 The investigator should not implement any deviation from, or changes of, the protocol without agreement by the sponsor and prior review and documented approval/favorable opinion from the IRB/IEC of an amendment, except where necessary to eliminate an immediate hazard(s) to trial subjects, or when the change(s) involves only logistical or administrative aspects of the trial (e.g., change in monitor(s), change of telephone number(s)). 4.5.3 The investigator, or person designated by the investigator, should document and explain any deviation from the approved protocol. 4.5.4 The investigator may implement a deviation from, or a change in, the protocol to eliminate an immediate hazard(s) to trial subjects without prior IRB/IEC approval/favorable opinion.
ICH E3, section 9.6
The sponsor should describe the quality management approach implemented in the trial and summarize important deviations from the predefined quality tolerance limits and remedial actions taken in the clinical study report
21CFR 312.53(vi) (a)
investigators selected “Will conduct the study(ies) in accordance with the relevant, current protocol(s) and will only make changes in a protocol after notifying the sponsor, except when necessary to protect the safety, the rights, or welfare of subjects.”
21CFR 56.108(a)
IRB shall….ensur[e] that changes in approved research….may not be initiated without IRB review and approval except where necessary to eliminate apparent immediate hazards to the human subjects.
21 CFR 56.108(b)
“IRB shall….follow written procedures for ensuring prompt reporting to the IRB, appropriate institutional officials, and the Food and Drug Administration of… any unanticipated problems involving risks to human subjects or others…[or] any instance of serious or continuing noncompliance with these regulations or the requirements or determinations of the IRB.”
45 CFR 46.103(b)(5)
Assurances applicable to federally supported or conducted research shall at a minimum include….written procedures for ensuring prompt reporting to the IRB….[of] any unanticipated problems involving risks to subjects or others or any serious or continuing noncompliance with this policy or the requirements or determinations of the IRB.
FDA Form-1572 (Section 9)
lists the commitments the investigator is undertaking in signing the 1572 wherein the clinical investigator agrees “to conduct the study(ies) in accordance with the relevant, current protocol(s) and will only make changes in a protocol after notifying the sponsor, except when necessary to protect the safety, the rights, or welfare of subjects… [and] not to make any changes in the research without IRB approval, except where necessary to eliminate apparent immediate hazards to the human subjects.”
A few key regulations and guidances (not meant to be a comprehensive list)
How Protocol Deviations are Implemented
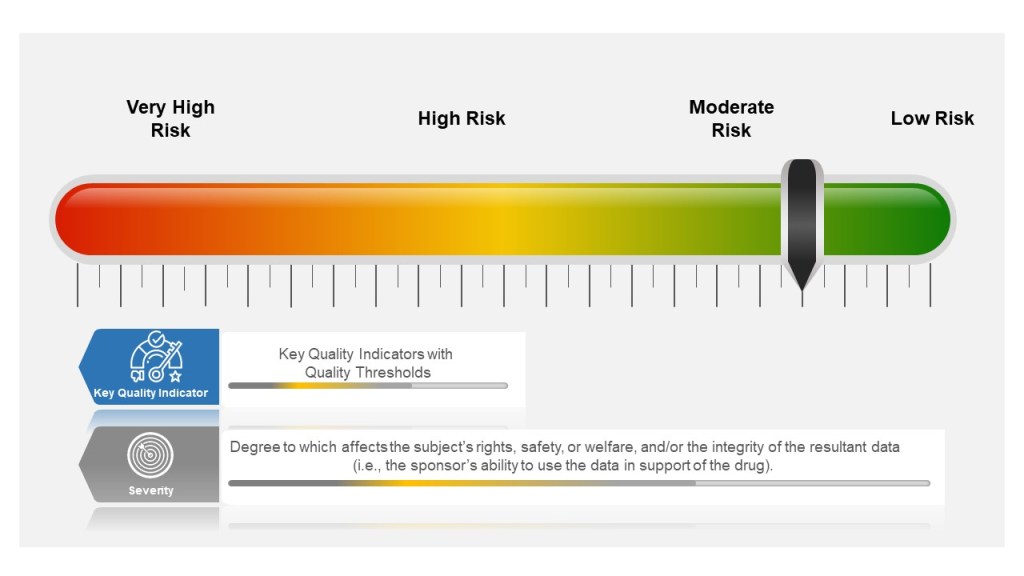
Many companies tend to have a failure scale built into their process, differentiating between protocol deviations and violations based on severity. Others use a minor, major, and even critical scale to denote differences in severity. The axis here for severity is the degree to which affects the subject’s rights, safety, or welfare, and/or the integrity of the resultant data (i.e., the sponsor’s ability to use the data in support of the drug).
Other companies divide into protocol deviations and violations:
Protocol Deviation: A protocol deviation occurs when, without significant consequences, the activities on a study diverge from the IRB-approved protocol, e.g., missing a visit window because the subject is traveling. Not as serious as a protocol violation.
Protocol Violation: A divergence from the protocol that materially (a) reduces the quality or completeness of the data, (b) makes the ICF inaccurate, or (c) impacts a subject’s safety, rights or welfare. Examples of protocol violations may include: inadequate or delinquent informed consent; inclusion/exclusion criteria not met; unreported SAEs; improper breaking of the blind; use of prohibited medication; incorrect or missing tests; mishandled samples; multiple visits missed or outside permissible windows; materially inadequate record-keeping; intentional deviation from protocol, GCP or regulations by study personnel; and subject repeated noncompliance with study requirements.
This is probably a place when nomenclature can serve to get in the way, rather than provide benefit. The EMA says pretty much the same in “ICH guideline E3 – questions and answers (R1).“
Principles of Events in Clinical Practice
Severity of the event is based on degree to which affects the subject’s rights, safety, or welfare, and/or the integrity of the resultant data
Events happen beyond the Protocol. These need to be managed appropriately as well.
The event needs to be categorized, evaluated and trended by the sponsor
Severity of the Event
Starting in the study planning stage, ICH E6(R2) GCP requires sponsors to identify risks to critical study processes and study data and to evaluate these risks based on likelihood, detectability and impact on subject safety and data integrity.
Sponsors then establish key quality indicators (KQIs) and quality tolerance thresholds. KQI is really just a key risk indicator and should be treated similarly.
Study events that exceed the risk threshold should trigger an evaluation to determine if action is needed. In this way, sponsors can proactively manage risk and address protocol noncompliance.
The best practice here is to have a living risk assessment for each study. Evaluate across studies to understand your overall organization risk, and look for opportunities for wide-scale mitigations. Feedup into your risk register.
Event Classification for Clinical Protocols and GCPs
Where the Event happens
Deviations in the clinical space are a great example of the management of supplier events, and at the end of the day there is little difference between a GMP supplier event management, a GLP or a GCP. The individual requirements might be different but the principles and the process are the same.
Each entity in the trial organization should have their own deviation system where they investigate deviations, performing root cause investigation and enacting CAPAs.
This is where it starts to get tricky. first of all, not all sites have the infrastructure to do this well. Second the nature of reporting, usually through the Electronic Data Capture (EDC) system, can lead to balkanization at the site. Site’s need to have strong compliance programs through compiling deviation details into a single sitewide system that allows the site to trend deviations across studies in addition to following sponsor reporting requirements.
Unfortunately too many site’s rely on the sponsor’s program. Sponsors need to be evaluating the strength of this program during site selection and through auditing.
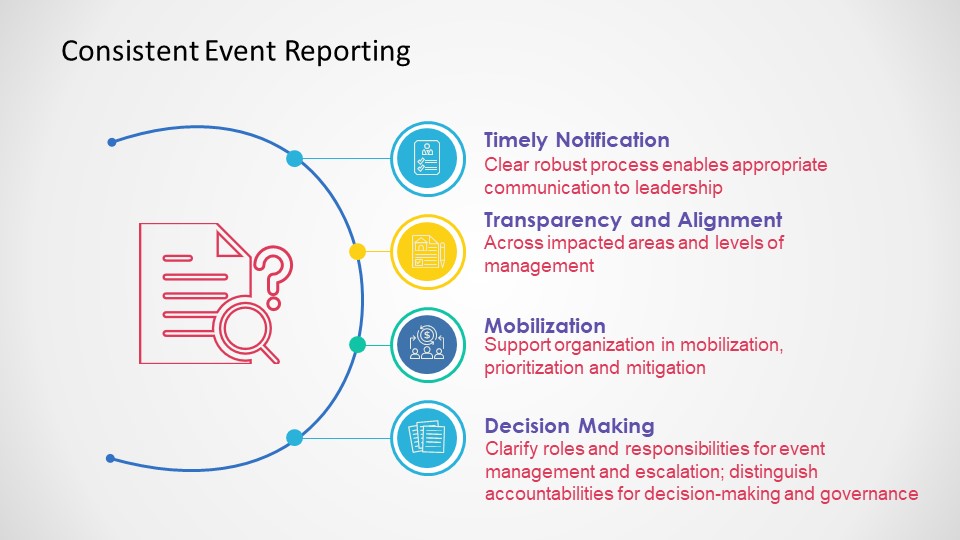
Events Happen
Consistent Event Reporting is Critical
Deviations should be to all process, procedure and plans, and just not the protocol.
Categorization and Trending
Categorizing deviations is usually a pain point and an area where more consistency needs to be driven. I recommend first having a good standard set of categorizations. The industry would benefit from adopting a standard, and I think Norman Goldfarb’s proposal is still the best.
Once you have categories, and understand to your KQIs and other aspects you need to make sure they are consistently done. The key mechanisms of this are: