We have an unfortunate habit of conflating regulatory process requirements with specific system functionality requirements. This confusion manifests most perversely in User Requirements Specifications that contain nebulous statements like “the system shall comply with 21 CFR Part 11” or “the system must meet EU GMP Annex 11 requirements.” These high-level regulatory citations represent a fundamental misunderstanding of what user requirements should accomplish and demonstrate a dangerous abdication of the detailed thinking required for effective validation.
The core problem is simple yet profound: lifecycle, risk management, and validation are organizational processes, not system characteristics. When we embed these process-level concepts into system requirements, we create validation exercises that test compliance theater rather than functional reality.
The Distinction That Changes Everything
User requirements specifications serve as the foundational document identifying what a system must do to meet specific business needs, product requirements, and operational constraints. They translate high-level business objectives into measurable, testable, and verifiable system behaviors. User requirements focus on what the system must accomplish, not how the organization manages its regulatory obligations.
Consider the fundamental difference between these approaches:
Problematic High-Level Requirement: “The system shall comply with 21 CFR Part 11 validation requirements.”
Proper Detailed Requirements:
“The system shall generate time-stamped audit trails for all data modifications, including user ID, date/time, old value, new value, and reason for change”
“The system shall enforce unique user identification through username/password combinations with minimum 8-character complexity requirements”
“The system shall prevent deletion of electronic records while maintaining complete audit trail visibility”
“The system shall provide electronic signature functionality that captures the printed name, date/time, and meaning of the signature”
The problematic version tells us nothing about what the system actually needs to do. The detailed versions provide clear, testable criteria that directly support Part 11 compliance while specifying actual system functionality.
Process vs. System: Understanding the Fundamental Categories
Lifecycle management, risk assessment, and validation represent organizational processes that exist independently of any specific system implementation. These processes define how an organization approaches system development, operation, and maintenance—they are not attributes that can be “built into” software.
Lifecycle processes encompass the entire journey from initial system conception through retirement, including stages such as requirements definition, design, development, testing, deployment, operation, and eventual decommissioning. A lifecycle approach ensures systematic progression through these stages with appropriate documentation, review points, and decision criteria. However, lifecycle management cannot be embedded as a system requirement because it describes the framework within which system development occurs, not system functionality itself.
Risk management processes involve systematic identification, assessment, and mitigation of potential hazards throughout system development and operation. Risk management influences system design decisions and validation approaches, but risk management itself is not a system capability—it is an organizational methodology for making informed decisions about system requirements and controls.
Validation processes establish documented evidence that systems consistently perform as intended and meet all specified requirements. Validation involves planning, execution, and documentation of testing activities, but validation is something done to systems, not something systems possess as an inherent characteristic.
The Illusion of Compliance Through Citation
When user requirements specifications contain broad regulatory citations rather than specific functional requirements, they create several critical problems that undermine effective validation:
Untestable Requirements: How does one verify that a system “complies with Part 11”? Such requirements provide no measurable criteria, no specific behaviors to test, and no clear success/failure conditions. Verification becomes a subjective exercise in regulatory interpretation rather than objective measurement of system performance.
Validation Theater: Broad compliance statements encourage checkbox validation exercises where teams demonstrate regulatory awareness without proving functional capability. These validations often consist of mapping system features to regulatory sections rather than demonstrating that specific user needs are met.
Scope Ambiguity: Part 11 and Annex 11 contain numerous requirements, many of which may not apply to specific systems or use cases. Blanket compliance statements fail to identify which specific regulatory requirements are relevant and which system functions address those requirements.
Change Management Nightmares: When requirements reference entire regulatory frameworks rather than specific system behaviors, any regulatory update potentially impacts system validation status. This creates unnecessary re-validation burdens and regulatory uncertainty.
Building Requirements That Actually Work
Effective user requirements specifications address regulatory compliance through detailed, system-specific functional requirements that directly support regulatory objectives. This approach ensures that validation activities test actual system capabilities rather than regulatory awareness
Focus on Critical Quality Attributes: Rather than citing broad compliance frameworks, identify the specific product and process attributes that regulatory requirements are designed to protect. For pharmaceutical systems, this might include data integrity, product traceability, batch genealogy, or contamination prevention.
Translate Regulatory Intent into System Functions: Understand what each applicable regulation is trying to achieve, then specify system behaviors that accomplish those objectives. Part 11’s audit trail requirements, for example, aim to ensure data integrity and accountability—translate this into specific system capabilities for logging, storing, and retrieving change records.
Maintain Regulatory Traceability: Document the relationship between specific system requirements and regulatory drivers, but do so through traceability matrices or design rationale documents rather than within the requirements themselves. This maintains clear regulatory justification while keeping requirements focused on system functionality.
Enable Risk-Based Validation: Detailed functional requirements support risk-based validation approaches by clearly identifying which system functions are critical to product quality, patient safety, or data integrity. This enables validation resources to focus on genuinely important capabilities rather than comprehensive regulatory coverage.
The Process-System Interface: Getting It Right
The relationship between organizational processes and system requirements should be managed through careful analysis and translation, not through broad regulatory citations. Effective user requirements development involves several critical steps:
Process Analysis: Begin by understanding the organizational processes that the system must support. This includes manufacturing processes, quality control workflows, regulatory reporting requirements, and compliance verification activities. However, the focus should be on what the system must enable, not how the organization manages compliance.
Regulatory Gap Analysis: Identify specific regulatory requirements that apply to the intended system use. Analyze these requirements to understand their functional implications for system design, but avoid copying regulatory language directly into system requirements.
Functional Translation: Convert regulatory requirements into specific, measurable system behaviors. This translation process requires deep understanding of both regulatory intent and system capabilities, but produces requirements that can be objectively verified.
Organizational Boundary Management: Clearly distinguish between requirements for system functionality and requirements for organizational processes. System requirements should focus exclusively on what the technology must accomplish, while process requirements address how the organization will use, maintain, and govern that technology.
Real-World Consequences of the Current Approach
The practice of embedding high-level regulatory requirements in user requirements specifications has created systemic problems throughout the pharmaceutical industry:
Validation Inefficiency: Teams spend enormous resources demonstrating broad regulatory compliance rather than proving that systems meet specific user needs. This misallocation of validation effort undermines both regulatory compliance and system effectiveness.
Inspection Vulnerability: When regulatory inspectors evaluate systems against broad compliance claims, they often identify gaps between high-level assertions and specific system capabilities. Detailed functional requirements provide much stronger inspection support by demonstrating specific regulatory compliance mechanisms.
System Modification Complexity: Changes to systems with broad regulatory requirements often trigger extensive re-validation activities, even when the changes don’t impact regulatory compliance. Specific functional requirements enable more targeted change impact assessments.
Cross-Functional Confusion: Development teams, validation engineers, and quality professionals often interpret broad regulatory requirements differently, leading to inconsistent implementation and validation approaches. Detailed functional requirements provide common understanding and clear success criteria.
A Path Forward: Detailed Requirements for Regulatory Success
The solution requires fundamental changes in how the pharmaceutical industry approaches user requirements development and regulatory compliance documentation:
Separate Compliance Strategy from System Requirements: Develop comprehensive regulatory compliance strategies that identify applicable requirements and define organizational approaches for meeting them, but keep these strategies distinct from system functional requirements. Use the compliance strategy to inform requirements development, not replace it.
Invest in Requirements Translation: Build organizational capability for translating regulatory requirements into specific, testable system functions. This requires regulatory expertise, system knowledge, and requirements engineering skills working together.
Implement Traceability Without Embedding: Maintain clear traceability between system requirements and regulatory drivers through external documentation rather than embedded citations. This preserves regulatory justification while keeping requirements focused on system functionality.
Focus Validation on Function: Design validation approaches that test system capabilities directly rather than compliance assertions. This produces stronger regulatory evidence while ensuring system effectiveness.
Lifecycle, risk management, and validation are organizational processes that guide how we develop and maintain systems—they are not system requirements themselves. When we treat them as such, we undermine both regulatory compliance and system effectiveness. The time has come to abandon this regulatory red herring and embrace requirements practices worthy of the products and patients we serve.
Writing user requirements that simply state “the system shall meet FDA 21 CFR Part 11 requirements” or “the system shall comply with EU Annex 11” is fundamentally bad practice. These high-level regulatory statements create ambiguity, shift responsibility inappropriately, and fail to provide the specific, testable criteria that effective requirements demand.
The Core Problem: Regulatory References Aren’t Technical Requirements
User requirements must be specific, measurable, and testable. When we write “the system shall meet Annex 11 and Part 11 requirements,” we’re not writing a technical requirement at all—we’re writing a reference to a collection of regulatory provisions that may or may not apply to our specific system context. This creates several fundamental problems that undermine the entire validation and verification process.
The most critical issue is ambiguity of technical scope. Annex 11 and Part 11 contains numerous provisions, but not all apply to every system. Some provisions address closed systems, others open systems. Some apply only when electronic records replace paper records, others when organizations rely on electronic records to perform regulated activities. Without specifying which technical provisions apply and how they translate into system functionality, we leave interpretation to individual team members—a recipe for inconsistent implementation.
Technical verification becomes impossible with such high-level statements. How does a tester verify that a system “meets Part 11”? They would need to become regulatory experts, interpret which provisions apply, translate those into testable criteria, and then execute tests—work that should have been done during requirements definition. This shifting of analytical responsibility from requirements authors to testers violates fundamental engineering principles.
Why This Happens: The Path of Least Resistance
The temptation to write high-level regulatory requirements stems from several understandable but misguided motivations. Requirements authors often lack deep regulatory knowledge and find it easier to reference entire regulations rather than analyze which specific technical provisions apply to their system. This approach appears comprehensive while avoiding the detailed work of regulatory interpretation.
Time pressure exacerbates this tendency. Writing “meet Part 11” takes minutes; properly analyzing regulatory requirements, determining technical applicability, and translating them into specific, testable statements takes days or weeks. Under project pressure, teams often choose the quick path without considering downstream consequences.
There’s also a false sense of completeness. Referencing entire regulations gives the impression of thorough coverage when it actually provides no technical coverage at all. It’s the requirements equivalent of writing “the system shall work properly”, technically correct but utterly useless for implementation or testing purposes.
Better Approach: Technical User Requirements
Effective regulatory user requirements break down high-level regulatory concepts into specific, measurable technical statements that directly address system functionality. Rather than saying “meet Part 11,” we need requirements that specify exactly what the system must do technically.
Access Control Requirements
Instead of: “The system shall meet Part 11 access control requirements”
Write:
“The system shall authenticate users through unique user ID and password combinations, where each combination is assigned to only one individual”
“The system shall automatically lock user sessions after 30 minutes of inactivity and require re-authentication”
“The system shall maintain an electronic log of all user authentication attempts, including failed attempts, with timestamps and user identifiers”
Electronic Record Generation Requirements
Instead of: “The system shall generate Part 11 compliant electronic records”
Write:
“The system shall generate electronic records that include all data required by the predicate rule, with no omission of required information”
“The system shall time-stamp all electronic records using computer-generated timestamps that cannot be altered by system users”
“The system shall detect and flag any unauthorized alterations to electronic records through checksum validation”
Audit Trail Requirements
Instead of: “The system shall maintain Part 11 compliant audit trails”
Write:
“The system shall automatically record the user ID, date, time, and type of action for all data creation, modification, and deletion operations”
“The system shall store audit trail records in a format that prevents user modification or deletion”
“The system shall provide audit trail search and filter capabilities by user, date range, and record type”
Electronic Signature Requirements
Instead of: “The system shall support Part 11 electronic signatures”
Write:
“Electronic signature records shall include the signer’s printed name, date and time of signing, and the purpose of the signature”
“The system shall verify signer identity through authentication requiring both user ID and password before accepting electronic signatures“
“The system shall cryptographically link electronic signatures to their associated records to prevent signature transfer or copying”
Annex 11 Technical Examples
EU Annex 11 requires similar technical specificity but with some European-specific nuances. Here are better technical requirement examples:
System Security Requirements
Instead of: “The system shall meet Annex 11 security requirements”
Write:
“The system shall implement role-based access control where user privileges are assigned based on documented job responsibilities”
“The system shall encrypt all data transmission between system components using AES 256-bit encryption”
“The system shall maintain user session logs that record login time, logout time, and all system functions accessed during each session”
Data Integrity Requirements
Instead of: “The system shall ensure Annex 11 data integrity”
Write:
“The system shall implement automated backup procedures that create complete system backups daily and verify backup integrity”
“The system shall prevent simultaneous modification of the same record by multiple users through record locking mechanisms”
“The system shall maintain original raw data in unalterable format while allowing authorized users to add comments or corrections with full audit trails”
System Change Control Requirements
Instead of: “The system shall implement Annex 11 change control”
Write:
“The system shall require authorized approval through electronic workflow before implementing any configuration changes that affect GMP functionality”
“The system shall maintain a complete history of all system configuration changes including change rationale, approval records, and implementation dates”
“The system shall provide the ability to revert system configuration to any previous approved state through documented rollback procedures”
The Business Case for Technical Requirements
Technical requirements save time and money. While writing detailed requirements requires more upfront effort, it prevents costly downstream problems. Clear technical requirements eliminate the need for interpretation during design, reduce testing iterations, and prevent regulatory findings during inspections.
Technical traceability becomes meaningful when requirements are specific. We can trace from business needs through technical specifications to test cases and validation results. This traceability is essential for regulatory compliance and change control.
System quality improves systematically when everyone understands exactly what technical functionality needs to be built and tested. Vague requirements lead to assumption-driven development where different team members make different assumptions about what’s technically needed.
Implementation Strategy for Technical Requirements
Start by conducting regulatory requirement analysis as a separate technical activity before writing user requirements. Identify which regulatory provisions apply to your specific system and translate them into technical functionality. Document this analysis and use it as the foundation for technical requirement writing.
Engage both regulatory and technical experts early in the requirements process. Don’t expect requirements authors to become overnight regulatory experts, but do ensure they have access to both regulatory knowledge and technical understanding when translating regulatory concepts into system requirements.
Use technical requirement templates that capture the essential technical elements of common regulatory requirements. This ensures consistency across projects and reduces the analytical burden on individual requirements authors.
Review requirements for technical testability. Every requirement should have an obvious technical verification method. If you can’t immediately see how to test a requirement technically, it needs to be rewritten.
Technical Requirements That Actually Work
High-level regulatory references have no place in technical user requirements documents. They create technical ambiguity where clarity is needed, shift analytical work to inappropriate roles, and fail to provide the specific technical guidance necessary for successful system implementation.
Better technical requirements translate regulatory concepts into specific, measurable, testable statements that directly address system technical functionality. This approach requires more upfront effort but delivers better technical outcomes: clearer system designs, more efficient testing, stronger regulatory compliance, and systems that actually meet user technical needs.
The pharmaceutical industry has matured beyond accepting “it must be compliant” as adequate technical guidance. Our technical requirements must mature as well, providing the specific, actionable technical direction that modern development teams need to build quality systems that truly serve patients and regulatory expectations.
As I’ve emphasized in previous posts about crafting good user requirements and building FUSE(P) user requirements, technical specificity and testability remain the hallmarks of effective requirements writing. Regulatory compliance requirements demand this same technical rigor—perhaps more so, given the patient safety implications of getting the technical implementation wrong.
The recently released draft of ICH Q3E addresses a critical gap that has persisted in pharmaceutical regulation for over two decades. Since the FDA’s 1999 Container Closure Systems guidance and the EMA’s 2005 Plastic Immediate Packaging Materials guideline, the regulatory landscape for extractables and leachables has remained fragmented across regions and dosage forms. This fragmentation has created significant challenges for global pharmaceutical companies, leading to inconsistent approaches, variable interpretation of requirements, and substantial regulatory uncertainty that ultimately impacts patient access to medicines.
The ICH Q3E guideline emerges from recognition that modern pharmaceutical development increasingly relies on complex drug-device combinations, novel delivery systems, and sophisticated manufacturing technologies that transcend traditional regulatory boundaries. Biologics, cell and gene therapies, combination products, and single-use manufacturing systems have created E&L challenges that existing guidance documents were never designed to address. The guideline’s comprehensive scope encompasses chemical entities, biologics, biotechnological products, and drug-device combinations across all dosage forms, establishing a unified framework that reflects the reality of contemporary pharmaceutical manufacturing.
The harmonization achieved through ICH Q3E extends beyond mere procedural alignment to establish fundamental scientific principles that can be applied consistently regardless of geographical location or specific regulatory jurisdiction. This represents a significant evolution from the current patchwork of guidance documents, each with distinct requirements and safety thresholds that often conflict or create unnecessary redundancy in global development programs.
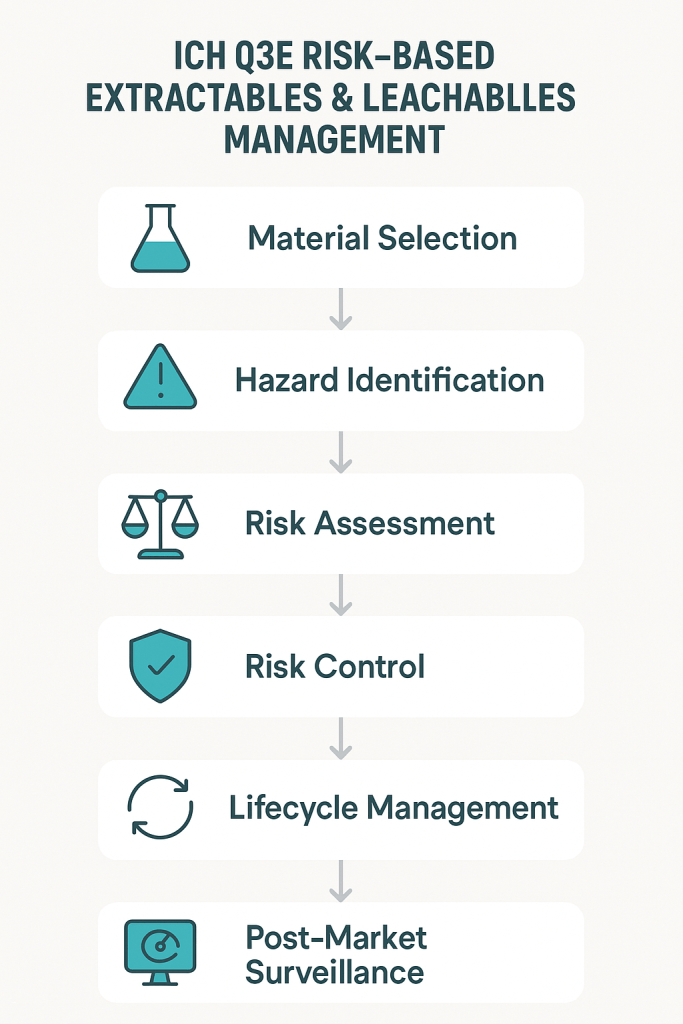
The most transformative aspect of ICH Q3E lies in its integration of comprehensive risk management principles derived from ICH Q9 throughout the entire E&L assessment process. This represents a fundamental departure from the prescriptive, one-size-fits-all approaches that have characterized previous guidance documents. The risk management framework encompasses four critical stages: hazard identification, risk assessment, risk control, and lifecycle management.
The hazard identification phase requires systematic evaluation of all materials of construction, manufacturing processes, and storage conditions that could contribute to extractables formation or leachables migration. This includes not only primary packaging components but also manufacturing equipment, single-use systems, filters, tubing, and any other materials that contact the drug substance or drug product during production, storage, or administration. The guideline recognizes that modern pharmaceutical manufacturing involves complex material interactions that require comprehensive evaluation beyond traditional container-closure system assessments.
Risk assessment under ICH Q3E employs a multi-dimensional approach that considers both the probability of extractables/leachables occurrence and the potential impact on product quality and patient safety. This assessment integrates factors such as contact time, temperature, pH, chemical compatibility, route of administration, patient population, and treatment duration. The framework explicitly acknowledges that risk varies significantly across different scenarios and requires tailored approaches rather than uniform requirements.
The risk control strategies outlined in ICH Q3E provide multiple pathways for managing identified risks, including material selection optimization, process parameter control, analytical monitoring, and specification limits. This flexibility enables pharmaceutical companies to develop cost-effective control strategies that are proportionate to the actual risks identified rather than applying maximum controls uniformly across all situations.
Lifecycle management ensures that E&L considerations remain integrated throughout product development, commercialization, and post-market surveillance. This includes provisions for managing material changes, process modifications, and the incorporation of new scientific knowledge as it becomes available. The lifecycle approach recognizes that E&L assessment is not a one-time activity but an ongoing process that must evolve with the product and available scientific understanding.
Safety Threshold Harmonization
ICH Q3E introduces a sophisticated threshold framework that harmonizes and extends the safety assessment principles developed through industry initiatives while addressing critical gaps in current approaches. The guideline establishes a risk-based threshold system that considers both mutagenic and non-mutagenic compounds while providing clear decision-making criteria for safety assessment.
For mutagenic compounds, ICH Q3E adopts a Threshold of Toxicological Concern (TTC) approach aligned with ICH M7 principles, establishing 1.5 μg/day as the default threshold for compounds with mutagenic potential. This represents harmonization with existing approaches while extending application to extractables and leachables that was previously addressed only through analogy or extrapolation.
For non-mutagenic compounds, the guideline introduces a tiered threshold system that considers route of administration, treatment duration, and patient population. The Safety Concern Threshold (SCT) varies based on these factors, with more conservative thresholds applied to high-risk scenarios such as parenteral administration or pediatric populations. This approach represents a significant advancement over current practice, which often applies uniform thresholds regardless of actual exposure scenarios or patient risk factors.
The Analytical Evaluation Threshold (AET) calculation methodology has been standardized and refined to provide consistent application across different analytical techniques and product configurations. The AET serves as the practical threshold for analytical identification and reporting, incorporating analytical uncertainty factors that ensure appropriate sensitivity for detecting compounds of potential safety concern.
The qualification threshold framework establishes clear decision points for when additional toxicological evaluation is required, reducing uncertainty and providing predictable pathways for safety assessment. Compounds below the SCT require no additional evaluation unless structural alerts are present, while compounds above the qualification threshold require comprehensive toxicological assessment using established methodologies.
Advanced Analytical Methodology Requirements
ICH Q3E establishes sophisticated analytical requirements that reflect advances in analytical chemistry and the increasing complexity of pharmaceutical products and manufacturing systems. The guideline requires fit-for-purpose analytical methods that are appropriately validated for their intended use, with particular emphasis on method capability to detect and quantify compounds at relevant safety thresholds.
The extraction study requirements have been standardized to ensure consistent generation of extractables profiles while allowing flexibility for product-specific optimization. The guideline establishes principles for solvent selection, extraction conditions, and extraction ratios that provide meaningful worst-case scenarios without introducing artifacts or irrelevant compounds. This standardization addresses a major source of variability in current practice, where different companies often use dramatically different extraction conditions that produce incomparable results.
Leachables assessment requirements emphasize the need for methods capable of detecting both known and unknown compounds in complex product matrices. The guideline recognizes the challenges associated with detecting low-level leachables in pharmaceutical formulations and provides guidance on method development strategies, including the use of placebo formulations, matrix subtraction approaches, and accelerated testing conditions that enhance detection capability.
The analytical uncertainty framework provides specific guidance on incorporating analytical variability into safety assessments, ensuring that measurement uncertainty does not compromise patient safety. This includes requirements for response factor databases, analytical uncertainty calculations, and the application of appropriate safety factors that account for analytical limitations.
Method validation requirements are tailored to the specific challenges of E&L analysis, including considerations for selectivity in complex matrices, detection limit requirements based on safety thresholds, and precision requirements that support reliable safety assessment. The guideline acknowledges that traditional pharmaceutical analytical validation approaches may not be directly applicable to E&L analysis and provides modified requirements that reflect the unique challenges of this application.
Material Science Integration and Innovation
ICH Q3E represents a significant advancement in the integration of material science principles into pharmaceutical quality systems. The guideline requires comprehensive material characterization that goes beyond simple compositional analysis to include understanding of manufacturing processes, potential degradation pathways, and interaction mechanisms that could lead to extractables formation.
The material selection guidance emphasizes proactive risk assessment during early development stages, enabling pharmaceutical companies to make informed material choices that minimize E&L risks rather than simply characterizing risks after materials have been selected. This approach aligns with Quality by Design principles and can significantly reduce development timelines and costs by avoiding late-stage material changes necessitated by unacceptable E&L profiles.
Single-use system assessment requirements reflect the increasing adoption of disposable manufacturing technologies in pharmaceutical production. The guideline provides specific frameworks for evaluating complex single-use assemblies that may contain multiple materials of construction and require additive risk assessment approaches. This addresses a critical gap in current guidance documents that were developed primarily for traditional reusable manufacturing equipment.
The guideline also addresses emerging materials and manufacturing technologies, including 3D-printed components, advanced polymer systems, and novel coating technologies. Provisions for evaluating innovative materials ensure that regulatory frameworks can accommodate technological advancement without compromising patient safety.
Comparison with Current Regulatory Frameworks
The transformative nature of ICH Q3E becomes evident when compared with existing regulatory approaches across different jurisdictions and application areas. The FDA’s 1999 Container Closure Systems guidance, while foundational, provides limited specific requirements and relies heavily on case-by-case assessment. This approach has led to significant variability in regulatory expectations and industry practice, creating uncertainty for both applicants and reviewers.
The EMA’s 2005 Plastic Immediate Packaging Materials guideline focuses specifically on plastic packaging materials and does not address the broader range of materials and applications covered by ICH Q3E. Additionally, the EMA guideline lacks specific safety thresholds, requiring product-specific risk assessment that can lead to inconsistent outcomes.
USP chapters <1663> and <1664> provide valuable technical guidance on extraction and leachables testing methodologies but do not establish safety thresholds or comprehensive risk assessment frameworks. These chapters serve as important technical references but require supplementation with safety assessment approaches from other sources.
The PQRI recommendations for orally inhaled and nasal drug products (OINDP) and parenteral and ophthalmic drug products (PODP) have provided industry-leading approaches to threshold-based safety assessment. However, these recommendations are limited to specific dosage forms and have not been formally adopted as regulatory requirements. ICH Q3E harmonizes and extends these approaches across all dosage forms while incorporating them into a formal regulatory framework.
Current European Pharmacopoeia requirements focus primarily on elemental extractables and do not address organic compounds comprehensively. The new EP chapter 2.4.35 on extractable elements represents an important advance but remains limited in scope compared to the comprehensive approach established by ICH Q3E.
ICH Q3E represents not merely an update or harmonization of existing approaches but a fundamental reconceptualization of E&L assessment that integrates the best elements of current practice while addressing critical gaps and inconsistencies.
Manufacturing Process Integration and Single-Use Systems
ICH Q3E places unprecedented emphasis on manufacturing process-related extractables and leachables, recognizing that modern pharmaceutical production increasingly relies on single-use systems, filters, tubing, and other disposable components that can contribute significantly to the overall E&L burden. This represents a major expansion from traditional container-closure system focus to encompass the entire manufacturing process.
The guideline establishes risk-based approaches for evaluating manufacturing equipment that consider factors such as contact time, process conditions, downstream processing steps, and the cumulative impact of multiple single-use components. This additive assessment approach acknowledges that even individually low-risk components can contribute to significant overall E&L levels when multiple components are used in series.
Single-use system assessment requirements address the complexity of modern bioprocessing equipment that may contain dozens of different materials of construction in a single assembly. The guideline provides frameworks for component-level assessment, assembly-level evaluation, and process-level integration that enable comprehensive risk assessment while maintaining practical feasibility.
The integration of manufacturing process E&L assessment with traditional container-closure system evaluation provides a holistic view of potential patient exposure that reflects the reality of modern pharmaceutical manufacturing. This comprehensive approach ensures that all sources of potential extractables and leachables are identified and appropriately controlled.
Biological Product Considerations and Specialized Applications
ICH Q3E provides specific considerations for biological products that reflect the unique challenges associated with protein stability, immunogenicity risk, and complex formulation requirements. Biological products often require specialized container-closure systems, delivery devices, and manufacturing processes that create distinct E&L challenges not adequately addressed by approaches developed for small molecule drugs.
The guideline addresses the potential for extractables and leachables to impact protein stability, aggregation, and biological activity through mechanisms that may not be captured by traditional chemical analytical approaches. This includes consideration of subvisible particle formation, protein adsorption, and catalytic degradation pathways that can be initiated by trace levels of extractables or leachables.
Immunogenicity considerations are explicitly addressed, recognizing that even very low levels of certain extractables or leachables could potentially trigger immune responses in sensitive patient populations. The guideline provides frameworks for assessing immunogenic risk that consider both the chemical nature of potential leachables and the clinical context of the biological product.
Cell and gene therapy applications receive special attention due to their unique manufacturing requirements, complex delivery systems, and often highly vulnerable patient populations. The guideline provides tailored approaches for these emerging therapeutic modalities that reflect their distinct risk profiles and manufacturing challenges.
Analytical Method Development and Validation Evolution
The analytical requirements established by ICH Q3E requires method capabilities that extend beyond traditional pharmaceutical analysis to encompass broad-spectrum unknown identification and quantification in complex matrices. This creates both challenges and opportunities for analytical laboratories and method development organizations.
Method development requirements emphasize systematic approaches to achieving required detection limits while maintaining selectivity in complex product matrices. The guideline provides specific guidance on extraction efficiency verification, matrix effect assessment, and the development of appropriate reference standards for quantification. These requirements ensure that analytical methods provide reliable data for safety assessment while maintaining practical feasibility.
Validation requirements are tailored to the unique challenges of E&L analysis, including considerations for compound identification confidence, quantification accuracy across diverse chemical structures, and method robustness across different product matrices. The guideline acknowledges that traditional pharmaceutical validation approaches may not be appropriate for E&L methods and provides modified requirements that reflect the specific challenges of this application.
The requirement for analytical uncertainty assessment and incorporation into safety evaluation represents a significant advancement in analytical quality assurance. Methods must not only provide accurate results but must also provide reliable estimates of measurement uncertainty that can be incorporated into risk assessment calculations.
Global Implementation Challenges and Opportunities
The implementation of ICH Q3E will require significant changes in pharmaceutical company practices, analytical capabilities, and regulatory review processes across all ICH regions. The comprehensive nature of the guideline means that virtually all pharmaceutical products will be impacted to some degree, creating both implementation challenges and opportunities for improved efficiency.
Training requirements will be substantial, as the guideline requires expertise in materials science, analytical chemistry, toxicology, and risk assessment that may not currently exist within all pharmaceutical organizations. The development of specialized E&L expertise will become increasingly important as companies seek to implement the guideline effectively.
Analytical infrastructure requirements may necessitate significant investments in instrumentation, method development capabilities, and reference standards. Smaller pharmaceutical companies may need to partner with specialized contract laboratories to access the required analytical capabilities.
Regulatory review processes will need to evolve to accommodate the risk-based approaches and comprehensive documentation requirements established by the guideline. Regulatory authorities will need to develop expertise in E&L assessment and establish consistent review practices across different therapeutic areas and product types.
The opportunities created by ICH Q3E implementation include improved regulatory predictability, reduced development timelines through early risk identification, and enhanced patient safety through more comprehensive E&L assessment. The harmonized approach should reduce the regulatory burden associated with multi-regional submissions while improving the overall quality of E&L assessments.
Future Evolution and Emerging Technologies
ICH Q3E has been designed with sufficient flexibility to accommodate emerging technologies and evolving scientific understanding. The risk-based framework can be adapted to new materials, manufacturing processes, and delivery systems as they are developed and implemented.
The guideline’s emphasis on scientific principles rather than prescriptive requirements enables adaptation to technological advances such as continuous manufacturing, advanced drug delivery systems, and personalized medicine approaches. This forward-looking design ensures that the guideline will remain relevant as pharmaceutical technology continues to evolve.
Provisions for incorporating new toxicological data and analytical methodologies ensure that the guideline can evolve with advancing scientific understanding. The lifecycle management approach enables updates and refinements based on accumulated experience and emerging scientific knowledge.
The integration with other ICH guidelines creates synergies that will facilitate future development of related guidance documents and ensure consistency across the broader ICH framework. This systematic approach to guideline development enhances the overall effectiveness of international pharmaceutical regulation.
Economic Impact and Industry Transformation
The implementation of ICH Q3E will have significant economic implications for the pharmaceutical industry, both in terms of implementation costs and long-term benefits. Initial implementation will require substantial investments in analytical capabilities, personnel training, and process modifications. However, the long-term benefits of harmonized requirements, improved regulatory predictability, and enhanced product quality are expected to provide significant value.
The harmonized approach should reduce the overall cost of global product development by eliminating duplicate testing requirements and reducing regulatory review timelines. Companies will be able to develop single global E&L strategies rather than maintaining multiple region-specific approaches.
Contract research organizations and analytical service providers will need to develop specialized capabilities to support pharmaceutical company implementation efforts. This will create new market opportunities while requiring significant investments in infrastructure and expertise.
The enhanced focus on risk-based assessment should enable more efficient allocation of resources to genuine safety concerns while reducing unnecessary testing and evaluation activities. This optimization of effort should improve overall industry efficiency while enhancing patient safety.
Patient Safety Enhancement and Risk Mitigation
The ultimate objective of ICH Q3E is enhanced patient safety through more comprehensive and scientifically rigorous assessment of extractables and leachables risks. The guideline achieves this objective through multiple mechanisms that address current gaps and limitations in E&L assessment practice.
The comprehensive material assessment requirements ensure that all potential sources of extractables and leachables are identified and evaluated. This includes not only traditional packaging materials but also manufacturing equipment, delivery device components, and any other materials that could contribute to patient exposure.
The harmonized safety threshold framework provides consistent and scientifically defensible criteria for safety assessment across all product types and administration routes. This eliminates the variability and uncertainty that can arise from inconsistent threshold application in current practice.
The risk-based approach enables appropriate allocation of assessment effort to genuine safety concerns while avoiding unnecessary evaluation of trivial risks. This optimization ensures that resources are focused on protecting patient safety rather than simply meeting regulatory requirements.
The lifecycle management requirements ensure that E&L considerations remain current throughout product development and commercialization. This ongoing attention to E&L issues helps identify and address emerging risks that might not be apparent during initial assessment.
Conclusion
ICH Q3E represents far more than an incremental improvement in extractables and leachables guidance; it establishes a new paradigm for pharmaceutical quality assurance that integrates materials science, analytical chemistry, toxicology, and risk management into a comprehensive framework that reflects the complexity of modern pharmaceutical development and manufacturing.
The guideline’s emphasis on scientific principles over prescriptive requirements creates a flexible framework that can accommodate the diverse and evolving landscape of pharmaceutical products while maintaining rigorous safety standards. This approach represents a significant maturation of regulatory science that moves beyond one-size-fits-all requirements to embrace risk-based, scientifically defensible assessment approaches.
The global harmonization achieved through ICH Q3E addresses one of the most significant challenges facing the pharmaceutical industry by providing consistent requirements and expectations across all major regulatory jurisdictions. This harmonization will facilitate more efficient global product development while enhancing patient safety through improved assessment practices.
The comprehensive scope of ICH Q3E ensures that extractables and leachables assessment evolves from a specialized concern for specific dosage forms to an integral component of pharmaceutical quality assurance across all products and therapeutic modalities. This integration reflects the reality that E&L considerations impact virtually all pharmaceutical products and must be systematically addressed throughout development and commercialization.
As the pharmaceutical industry prepares for ICH Q3E implementation, the focus must be on building the scientific expertise, analytical capabilities, and quality systems necessary to realize the guideline’s potential for enhancing patient safety while improving development efficiency. The successful implementation of ICH Q3E will mark a new era in pharmaceutical quality assurance that better serves patients, regulators, and the pharmaceutical industry through more rigorous, consistent, and scientifically defensible approaches to extractables and leachables assessment.
The transformation initiated by ICH Q3E extends beyond technical requirements to encompass fundamental changes in how pharmaceutical companies approach material selection, process design, analytical strategy, and risk management. This holistic transformation will ultimately deliver safer, higher-quality pharmaceutical products to patients worldwide while establishing a more efficient and predictable regulatory environment that facilitates innovation and global access to medicines.
The draft revision of EU GMP Chapter 4 introduces what can only be described as a revolutionary framework for data governance systems. This isn’t merely an update to existing documentation requirements—it is a keystone document that cements the decade long paradigm shift of data governance as the cornerstone of modern pharmaceutical quality systems.
The Genesis of Systematic Data Governance
The most striking aspect of the draft Chapter 4 is the introduction of sections 4.10 through 4.18, which establish data governance systems as mandatory infrastructure within pharmaceutical quality systems. This comprehensive framework emerges from lessons learned during the past decade of data integrity enforcement actions and reflects the reality that modern pharmaceutical manufacturing operates in an increasingly digital environment where traditional documentation approaches are insufficient.
The requirement that regulated users “establish a data governance system integral to the pharmaceutical quality system” moves far beyond the current Chapter 4’s basic documentation requirements. This integration ensures that data governance isn’t treated as an IT afterthought or compliance checkbox, but rather as a fundamental component of how pharmaceutical companies ensure product quality and patient safety. The emphasis on integration with existing pharmaceutical quality systems builds on synergies that I’ve previously discussed in my analysis of how data governance, data quality, and data integrity work together as interconnected pillars.
The requirement for regular documentation and review of data governance arrangements establishes accountability and ensures continuous improvement. This aligns with my observations about risk-based thinking where effective quality systems must anticipate, monitor, respond, and learn from their operational environment.
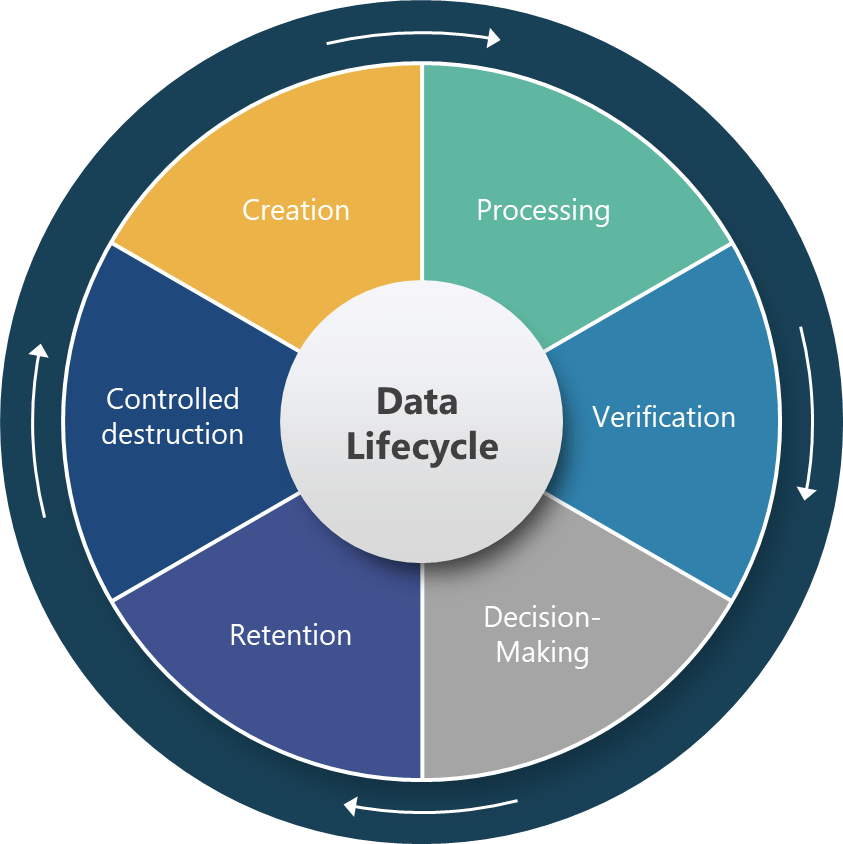
Comprehensive Data Lifecycle Management
Section 4.12 represents perhaps the most technically sophisticated requirement in the draft, establishing a six-stage data lifecycle framework that covers creation, processing, verification, decision-making, retention, and controlled destruction. This approach acknowledges that data integrity cannot be ensured through point-in-time controls but requires systematic management throughout the entire data journey.
The specific requirement for “reconstruction of all data processing activities” for derived data establishes unprecedented expectations for data traceability and transparency. This requirement will fundamentally change how pharmaceutical companies design their data processing workflows, particularly in areas like process analytical technology (PAT), manufacturing execution systems (MES), and automated batch release systems where raw data undergoes significant transformation before supporting critical quality decisions.
The lifecycle approach also creates direct connections to computerized system validation requirements under Annex 11, as noted in section 4.22. This integration ensures that data governance systems are not separate from, but deeply integrated with, the technical systems that create, process, and store pharmaceutical data. As I’ve discussed in my analysis of computer system validation frameworks, effective validation programs must consider the entire system ecosystem, not just individual software applications.
Risk-Based Data Criticality Assessment
The draft introduces a sophisticated two-dimensional risk assessment framework through section 4.13, requiring organizations to evaluate both data criticality and data risk. Data criticality focuses on the impact to decision-making and product quality, while data risk considers the opportunity for alteration or deletion and the likelihood of detection. This framework provides a scientific basis for prioritizing data protection efforts and designing appropriate controls.
This approach represents a significant evolution from current practices where data integrity controls are often applied uniformly regardless of the actual risk or impact of specific data elements. The risk-based framework allows organizations to focus their most intensive controls on the data that matters most while applying appropriate but proportionate controls to lower-risk information. This aligns with principles I’ve discussed regarding quality risk management under ICH Q9(R1), where structured, science-based approaches reduce subjectivity and improve decision-making.
The requirement to assess “likelihood of detection” introduces a crucial element often missing from traditional data integrity approaches. Organizations must evaluate not only how to prevent data integrity failures but also how quickly and reliably they can detect failures that occur despite preventive controls. This assessment drives requirements for monitoring systems, audit trail analysis capabilities, and incident detection procedures.
Service Provider Oversight and Accountability
Section 4.18 establishes specific requirements for overseeing service providers’ data management policies and risk control strategies. This requirement acknowledges the reality that modern pharmaceutical operations depend heavily on cloud services, SaaS platforms, contract manufacturing organizations, and other external providers whose data management practices directly impact pharmaceutical company compliance.
The risk-based frequency requirement for service provider reviews represents a practical approach that allows organizations to focus oversight efforts where they matter most while ensuring that all service providers receive appropriate attention. For more details on the evolving regulatory expectations around supplier management see the post “draft Annex 11’s supplier oversight requirements“.
The service provider oversight requirement also creates accountability throughout the pharmaceutical supply chain, ensuring that data integrity expectations extend beyond the pharmaceutical company’s direct operations to encompass all entities that handle GMP-relevant data. This approach recognizes that regulatory accountability cannot be transferred to external providers, even when specific activities are outsourced.
Operational Implementation Challenges
The transition to mandatory data governance systems will present significant operational challenges for most pharmaceutical organizations. The requirement for “suitably designed systems, the use of technologies and data security measures, combined with specific expertise” in section 4.14 acknowledges that effective data governance requires both technological infrastructure and human expertise.
Organizations will need to invest in personnel with specialized data governance expertise, implement technology systems capable of supporting comprehensive data lifecycle management, and develop procedures for managing the complex interactions between data governance requirements and existing quality systems. This represents a substantial change management challenge that will require executive commitment and cross-functional collaboration.
The requirement for regular review of risk mitigation effectiveness in section 4.17 establishes data governance as a continuous improvement discipline rather than a one-time implementation project. Organizations must develop capabilities for monitoring the performance of their data governance systems and adjusting controls as risks evolve or new technologies are implemented.
The integration with quality risk management principles throughout sections 4.10-4.22 creates powerful synergies between traditional pharmaceutical quality systems and modern data management practices. This integration ensures that data governance supports rather than competes with existing quality initiatives while providing a systematic framework for managing the increasing complexity of pharmaceutical data environments.
The draft’s emphasis on data ownership throughout the lifecycle in section 4.15 establishes clear accountability that will help organizations avoid the diffusion of responsibility that often undermines data integrity initiatives. Clear ownership models provide the foundation for effective governance, accountability, and continuous improvement.
Barriers, or controls, are one of the fundamental elements of root cause analysis. By understanding barriers—including their types and functions—we can understand both why a problem happened and how it can be prevented in the future. An evaluation of current process controls as part of root cause analysis can help determine whether all the current barriers pertaining to the problem you are investigating were present and effective.
Understanding Barrier Analysis
At its simplest, barrier analysis is a three-part brainstorm that examines the status and effectiveness of safety measures:
Barrier Analysis
Barriers that failed
Barriers that were not used
Barriers that did not exist
The key to this brainstorming session is to try to find all of the failed, unused, or nonexistent barriers. Do not be concerned if you are not certain which category they belong in initially.
Types of Barriers: Technical, Human, and Organizational
Most forms of barrier analysis examine two primary types: technical and administrative. Administrative barriers can be further broken down into “human” and “organizational” categories.
Choose
Technical
Human
Organizational
If
A technical or engineering control exists
The control relies on a human reviewer or operator
The control involves a transfer of responsibility. For example, a document reviewed by both manufacturing and quality.
Examples
Separation among manufacturing or packaging lines Emergency power supply Dedicated equipment Barcoding Keypad controlled doors Separated storage for components Software that prevents a workflow from going further if a field is not completed Redundant designs
Training and certifications Use of checklist Verification of critical task by a second person
Clear procedures and policies Adequate supervision Adequate load of work Periodic process audits
Preventive vs. Mitigative Barriers: A Critical Distinction
A fundamental aspect of barrier analysis involves understanding the difference between preventive and mitigative barriers. This distinction is crucial for comprehensive risk management and aligns with widely used frameworks such as bow-tie analysis.
Preventive Barriers
Preventive barriers are measures designed to prevent the top event from occurring. These barriers:
Focus on stopping incidents before they happen
Act as the first line of defense against threats
Aim to reduce the likelihood that a risk will materialize
Are proactive in nature, addressing potential causes before they can lead to unwanted events
Examples of preventive barriers include:
Regular equipment maintenance programs
Training and certification programs
Access controls and authentication systems
Equipment qualification protocols (IQ/OQ/PQ) validating proper installation and operation
Mitigative Barriers
Mitigative barriers are designed to reduce the impact and severity of consequences after the top event has occurred. These barriers:
Focus on damage control rather than prevention
Act to minimize harm when preventive measures have failed
Reduce the severity or substantially decrease the likelihood of consequences occurring
Are reactive in nature, coming into play after a risk has materialized
Examples of mitigative barriers include:
Alarm systems and response procedures
Containment measures for hazards
Emergency response teams and protocols
Backup power systems for critical operations
Timeline and Implementation Differences
The timing of barrier implementation and failure differs significantly between preventive and mitigative barriers:
Preventive barriers often fail over days, weeks, or years before the top event occurs, providing more opportunities for identification and intervention
Mitigative barriers often fail over minutes or hours after the top event occurs, requiring higher reliability and immediate effectiveness
This timing difference leads to higher reliance on mitigative barriers working correctly the first time
Enhanced Barrier Analysis Framework
Building on the traditional three-part analysis, organizations should incorporate the preventive vs. mitigative distinction into their barrier evaluation:
Enhanced Barrier Analysis
Preventive barriers that failed
Preventive barriers that were not used
Preventive barriers that did not exist
Mitigative barriers that failed
Mitigative barriers that were not used
Mitigative barriers that did not exist
Integration with Risk Assessment
These barriers are the same as current controls in risk assessment, which is key in a wide variety of risk assessment tools. The optimal approach involves balancing both preventive and mitigative barriers without placing reliance on just one type. Some companies may favor prevention by placing high confidence in their systems and practices, while others may emphasize mitigation through reactive policies, but neither approach alone is advisable as they each result in over-reliance on one type of barrier.
Practical Application
When conducting barrier analysis as part of root cause investigation:
Identify all relevant barriers that were supposed to protect against the incident
Classify each barrier as preventive or mitigative based on its intended function
Determine the barrier type: technical, human, or organizational
Assess barrier status: failed, not used, or did not exist
Evaluate the balance between preventive and mitigative measures
Develop corrective actions that address gaps in both preventive and mitigative barriers
This comprehensive approach to barrier analysis provides a more nuanced understanding of how incidents occur and how they can be prevented or their consequences minimized in the future. By understanding both the preventive and mitigative functions of barriers, organizations can develop more robust risk management strategies that address threats at multiple points in the incident timeline.