Just as magpies are attracted to shiny objects, collecting them without purpose or pattern, professionals often find themselves drawn to the latest tools, techniques, or technologies that promise quick fixes or dramatic improvements. We attend conferences, read articles, participate in webinars, and invariably come away with new tools to add to our professional toolkit.

This approach typically manifests in several recognizable patterns. You might see a quality professional enthusiastically implementing a fishbone diagram after attending a workshop, only to abandon it a month later for a new problem-solving methodology learned in a webinar. Or you’ve witnessed a manager who insists on using a particular project management tool simply because it worked well in their previous organization, regardless of its fit for current challenges. Even more common is the organization that accumulates a patchwork of disconnected tools over time – FMEA here, 5S there, with perhaps some Six Sigma tools sprinkled throughout – without a coherent strategy binding them together.
The consequences of this unsystematic approach are far-reaching. Teams become confused by constantly changing methodologies. Organizations waste resources on tools that don’t address fundamental needs and fail to build coherent quality systems that sustainably drive improvement. Instead, they create what might appear impressive on the surface but is fundamentally an incoherent collection of disconnected tools and techniques.
As I discussed in my recent post on methodologies, frameworks, and tools, this haphazard approach represents a fundamental misunderstanding of how effective quality systems function. The solution isn’t simply to stop acquiring new tools but to be deliberate and systematic in evaluating, selecting, and implementing them by starting with frameworks – the conceptual scaffolding that provides structure and guidance for our quality efforts – and working methodically toward appropriate tool selection.
I will outline a path from frameworks to tools in this post, utilizing the document pyramid as a structural guide. We’ll examine how the principles of sound systems design can inform this journey, how coherence emerges from thoughtful alignment of frameworks and tools, and how maturity models can help us track our progress. By the end, you’ll have a clear roadmap for transforming your organization’s approach to tool selection from random collection to strategic implementation.
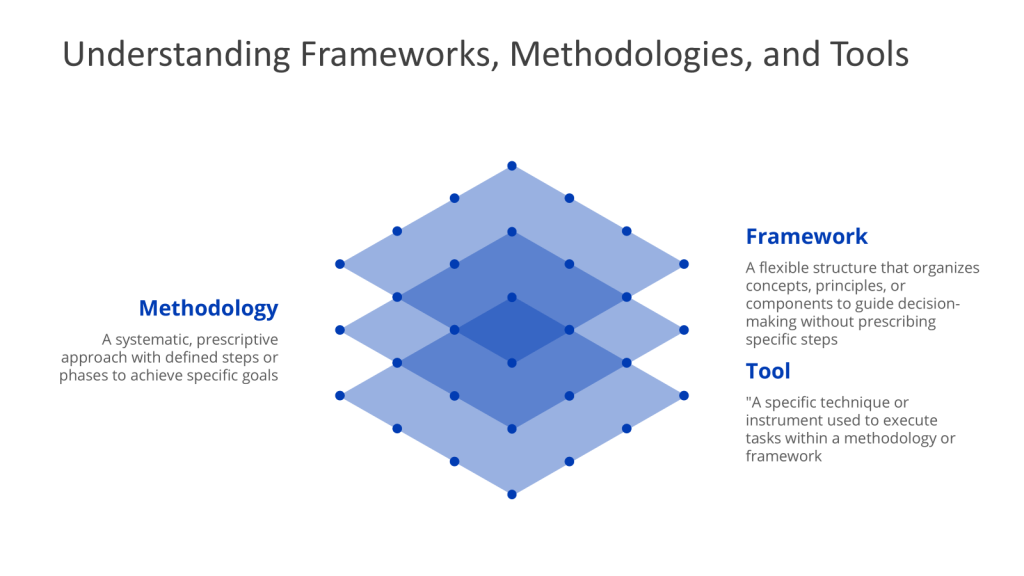
Understanding the Hierarchy: Frameworks, Methodologies, and Tools
Here is a brief refresher:
- A framework provides a flexible structure that organizes concepts, principles, and practices to guide decision-making. Unlike methodologies, frameworks are not rigidly sequential; they provide a mental model or lens through which problems can be analyzed. Frameworks emphasize what needs to be addressed rather than how to address it.
- A methodology is a systematic, step-by-step approach to solving problems or achieving objectives. It provides a structured sequence of actions, often grounded in theoretical principles, and defines how tasks should be executed. Methodologies are prescriptive, offering clear guidelines to ensure consistency and repeatability.
- A tool is a specific technique, model, or instrument used to execute tasks within a methodology or framework. Tools are action-oriented and often designed for a singular purpose, such as data collection, analysis, or visualization.
How They Interrelate: Building a Cohesive Strategy
The relationship between frameworks, methodologies, and tools is not merely hierarchical but interconnected and synergistic. A framework provides the conceptual structure for understanding a problem, the methodology defines the execution plan, and tools enable practical implementation.
To illustrate this integration, consider how these elements work together in various contexts:
In Systems Thinking:
- Framework: Systems theory identifies inputs, processes, outputs, and feedback loops
- Methodology: A 5-phase approach (problem structuring, dynamic modeling, scenario planning) guides analysis
- Tools: Causal loop diagrams map relationships; simulation software models system behavior
In Quality by Design (QbD):
- Framework: The ICH Q8 guideline outlines quality objectives
- Methodology: Define QTPP → Identify Critical Quality Attributes → Design experiments
- Tools: Design of Experiments (DoE) optimizes process parameters
Without frameworks, methodologies lack context and direction. Without methodologies, frameworks remain theoretical abstractions. Without tools, methodologies cannot be operationalized. The coherence and effectiveness of a quality management system depend on the proper alignment and integration of all three elements.
Understanding this hierarchy and interconnection is essential as we move toward establishing a deliberate path from frameworks to tools using the document pyramid structure.
The Document Pyramid: A Structure for Implementation
The document pyramid represents a hierarchical approach to organizing quality management documentation, which provides an excellent structure for mapping the path from frameworks to tools. In traditional quality systems, this pyramid typically consists of four levels: policies, procedures, work instructions, and records. However, I’ve found that adding an intermediate “program” level between policies and procedures creates a more effective bridge between high-level requirements and operational implementation.
Traditional Document Hierarchy in Quality Systems
Before examining the enhanced pyramid, let’s understand the traditional structure:
Policy Level: At the apex of the pyramid, policies establish the “what” – the requirements that must be met. They articulate the organization’s intentions, direction, and commitments regarding quality. Policies are typically broad, principle-based statements that apply across the organization.
Procedure Level: Procedures define the “who, what, when” of activities. They outline the sequence of steps, responsibilities, and timing for key processes. Procedures are more specific than policies but still focus on process flow rather than detailed execution.
Work Instruction Level: Work instructions provide the “how” – detailed steps for performing specific tasks. They offer step-by-step guidance for executing activities and are typically used by frontline staff directly performing the work.
Records Level: At the base of the pyramid, records provide evidence that work was performed according to requirements. They document the results of activities and serve as proof of compliance.
This structure establishes a logical flow from high-level requirements to detailed execution and documentation. However, in complex environments where requirements must be interpreted in various ways for different contexts, a gap often emerges between policies and procedures.
The Enhanced Pyramid: Adding the Program Level
To address this gap, I propose adding a “program” level between policies and procedures. The program level serves as a mapping requirement that shows the various ways to interpret high-level requirements for specific needs.
The beauty of the program document is that it helps translate from requirements (both internal and external) to processes and procedures. It explains how they interact and how they’re supported by technical assessments, risk management, and other control activities. Think of it as the design document and the connective tissue of your quality system.
With this enhanced structure, the document pyramid now consists of five levels:
- Policy Level (frameworks): Establishes what must be done
- Program Level (methodologies): Translates requirements into systems design
- Procedure Level: Defines who, what, when of activities
- Work Instruction Level (tools): Provides detailed how-to guidance
- Records Level: Evidences that activities were performed
This enhanced pyramid provides a clear structure for mapping our journey from frameworks to tools.

Mapping Frameworks, Methodologies, and Tools to the Document Pyramid
When we overlay our hierarchy of frameworks, methodologies, and tools onto the document pyramid, we can see the natural alignment:
Frameworks operate at the Policy Level. They establish the conceptual structure and principles that guide the entire quality system. Policies articulate the “what” of quality management, just as frameworks define the “what” that needs to be addressed.
Methodologies align with the Program Level. They translate the conceptual guidance of frameworks into systematic approaches for implementation. The program level provides the connective tissue between high-level requirements and operational processes, similar to how methodologies bridge conceptual frameworks and practical tools.
Tools correspond to the Work Instruction Level. They provide specific techniques for executing tasks, just as work instructions detail exactly how to perform activities. Both are concerned with practical, hands-on implementation.
The Procedure Level sits between methodologies and tools, providing the organizational structure and process flow that guide tool selection and application. Procedures define who will use which tools, when they will be used, and in what sequence.
Finally, Records provide evidence of proper tool application and effectiveness. They document the results achieved through the application of tools within the context of methodologies and frameworks.
This mapping provides a structural framework for our journey from high-level concepts to practical implementation. It helps ensure that tool selection is not arbitrary but rather guided by and aligned with the organization’s overall quality framework and methodology.
Systems Thinking as a Meta-Framework
To guide our journey from frameworks to tools, we need a meta-framework that provides overarching principles for system design and evaluation. Systems thinking offers such a meta-framework, and I believe we can apply eight key principles that can be applied across the document pyramid to ensure coherence and effectiveness in our quality management system.
The Eight Principles of Good Systems
These eight principles form the foundation of effective system design, regardless of the specific framework, methodology, or tools employed:
Balance
Definition: The system creates value for multiple stakeholders. While the ideal is to develop a design that maximizes value for all key stakeholders, designers often must compromise and balance the needs of various stakeholders.
Application across the pyramid:
- At the Policy/Framework level, balance ensures that quality objectives serve multiple organizational goals (compliance, customer satisfaction, operational efficiency)
- At the Program/Methodology level, balance guides the design of systems that address diverse stakeholder needs
- At the Work Instruction/Tool level, balance influences tool selection to ensure all stakeholder perspectives are considered
Congruence
Definition: The degree to which system components are aligned and consistent with each other and with other organizational systems, culture, plans, processes, information, resource decisions, and actions.
Application across the pyramid:
- At the Policy/Framework level, congruence ensures alignment between quality frameworks and organizational strategy
- At the Program/Methodology level, congruence guides the development of methodologies that integrate with existing systems
- At the Work Instruction/Tool level, congruence ensures selected tools complement rather than contradict each other
Convenience
Definition: The system is designed to be as convenient as possible for participants to implement (a.k.a. user-friendly). The system includes specific processes, procedures, and controls only when necessary.
Application across the pyramid:
- At the Policy/Framework level, convenience influences the selection of frameworks that suit organizational culture
- At the Program/Methodology level, convenience shapes methodologies to be practical and accessible
- At the Work Instruction/Tool level, convenience drives the selection of tools that users can easily adopt and apply
Coordination
Definition: System components are interconnected and harmonized with other (internal and external) components, systems, plans, processes, information, and resource decisions toward common action or effort. This goes beyond congruence and is achieved when individual components operate as a fully interconnected unit.
Application across the pyramid:
- At the Policy/Framework level, coordination ensures frameworks complement each other
- At the Program/Methodology level, coordination guides the development of methodologies that work together as an integrated system
- At the Work Instruction/Tool level, coordination ensures tools are compatible and support each other
Elegance
Definition: Complexity vs. benefit — the system includes only enough complexity as necessary to meet stakeholders’ needs. In other words, keep the design as simple as possible but no simpler while delivering the desired benefits.
Application across the pyramid:
- At the Policy/Framework level, elegance guides the selection of frameworks that provide sufficient but not excessive structure
- At the Program/Methodology level, elegance shapes methodologies to include only necessary steps
- At the Work Instruction/Tool level, elegance influences the selection of tools that solve problems without introducing unnecessary complexity
Human-Centered
Definition: Participants in the system are able to find joy, purpose, and meaning in their work.
Application across the pyramid:
- At the Policy/Framework level, human-centeredness ensures frameworks consider human factors
- At the Program/Methodology level, human-centeredness shapes methodologies to engage and empower participants
- At the Work Instruction/Tool level, human-centeredness drives the selection of tools that enhance rather than diminish human capabilities
Learning
Definition: Knowledge management, with opportunities for reflection and learning (learning loops), is designed into the system. Reflection and learning are built into the system at key points to encourage single- and double-loop learning from experience.
Application across the pyramid:
- At the Policy/Framework level, learning influences the selection of frameworks that promote improvement
- At the Program/Methodology level, learning shapes methodologies to include feedback mechanisms
- At the Work Instruction/Tool level, learning drives the selection of tools that generate insights and promote knowledge creation
Sustainability
Definition: The system effectively meets the near- and long-term needs of current stakeholders without compromising the ability of future generations of stakeholders to meet their own needs.
Application across the pyramid:
- At the Policy/Framework level, sustainability ensures frameworks consider long-term viability
- At the Program/Methodology level, sustainability shapes methodologies to create lasting value
- At the Work Instruction/Tool level, sustainability influences the selection of tools that provide enduring benefits
These eight principles serve as evaluation criteria throughout our journey from frameworks to tools. They help ensure that each level of the document pyramid contributes to a coherent, effective, and sustainable quality system.
Systems Thinking and the Five Key Questions
In addition to these eight principles, systems thinking guides us to ask five key questions that apply across the document pyramid:
- What is the purpose of the system? What happens in the system?
- What is the system? What’s inside? What’s outside? Set the boundaries, the internal elements, and elements of the system’s environment.
- What are the internal structure and dependencies?
- How does the system behave? What are the system’s emergent behaviors, and do we understand their causes and dynamics?
- What is the context? Usually in terms of bigger systems and interacting systems.
Answering these questions at each level of the document pyramid helps ensure alignment and coherence. For example:
- At the Policy/Framework level, we ask about the overall purpose of our quality system, its boundaries, and its context within the broader organization
- At the Program/Methodology level, we define the internal structure and dependencies of specific quality initiatives
- At the Work Instruction/Tool level, we examine how individual tools contribute to system behavior and objectives
By applying systems thinking principles and questions throughout our journey from frameworks to tools, we create a coherent quality system rather than a collection of disconnected elements.
Coherence in Quality Systems
Coherence goes beyond mere alignment or consistency. While alignment ensures that different elements point in the same direction, coherence creates a deeper harmony where components work together to produce emergent properties that transcend their individual contributions.
In quality systems, coherence means that our frameworks, methodologies, and tools don’t merely align on paper but actually work together organically to produce desired outcomes. The parts reinforce each other, creating a whole that is greater than the sum of its parts.
Building Coherence Through the Document Pyramid
The enhanced document pyramid provides an excellent structure for building coherence in quality systems. Each level must not only align with those above and below it but also contribute to the emergent properties of the whole system.
At the Policy/Framework level, coherence begins with selecting frameworks that complement each other and align with organizational context. For example, combining systems thinking with Quality by Design creates a more coherent foundation than either framework alone.
At the Program/Methodology level, coherence develops through methodologies that translate framework principles into practical approaches while maintaining their essential character. The program level is where we design systems that build order through their function rather than through rigid control.
At the Procedure level, coherence requires processes that flow naturally from methodologies while addressing practical organizational needs. Procedures should feel like natural expressions of higher-level principles rather than arbitrary rules.
At the Work Instruction/Tool level, coherence depends on selecting tools that embody the principles of chosen frameworks and methodologies. Tools should not merely execute tasks but reinforce the underlying philosophy of the quality system.
Throughout the pyramid, coherence is enhanced by using similar building blocks across systems. Risk management, data integrity, and knowledge management can serve as common elements that create consistency while allowing for adaptation to specific contexts.
The Framework-to-Tool Path: A Structured Approach
Building on the foundations we’ve established – the hierarchy of frameworks, methodologies, and tools; the enhanced document pyramid; systems thinking principles; and coherence concepts – we can now outline a structured approach for moving from frameworks to tools in a deliberate and coherent manner.
Step 1: Framework Selection Based on System Needs
The journey begins at the Policy level with the selection of appropriate frameworks. This selection should be guided by organizational context, strategic objectives, and the nature of the challenges being addressed.
Key considerations in framework selection include:
- System Purpose: What are we trying to achieve? Different frameworks emphasize different aspects of quality (e.g., risk reduction, customer satisfaction, operational excellence).
- System Context: What is our operating environment? Regulatory requirements, industry standards, and market conditions all influence framework selection.
- Stakeholder Needs: Whose interests must be served? Frameworks should balance the needs of various stakeholders, from customers and employees to regulators and shareholders.
- Organizational Culture: What approaches will resonate with our people? Frameworks should align with organizational values and ways of working.
Examples of quality frameworks include Systems Thinking, Quality by Design (QbD), Total Quality Management (TQM), and various ISO standards. Organizations often adopt multiple complementary frameworks to address different aspects of their quality system.
The output of this step is a clear articulation of the selected frameworks in policy documents that establish the conceptual foundation for all subsequent quality efforts.
Step 2: Translating Frameworks to Methodologies
At the Program level, we translate the selected frameworks into methodologies that provide systematic approaches for implementation. This translation occurs through program documents that serve as connective tissue between high-level principles and operational procedures.
Key activities in this step include:
- Framework Interpretation: How do our chosen frameworks apply to our specific context? Program documents explain how framework principles translate into organizational approaches.
- Methodology Selection: What systematic approaches will implement our frameworks? Examples include Six Sigma (DMAIC), 8D problem-solving, and various risk management methodologies.
- System Design: How will our methodologies work together as a coherent system? Program documents outline the interconnections and dependencies between different methodologies.
- Resource Allocation: What resources are needed to support these methodologies? Program documents identify the people, time, and tools required for successful implementation.
The output of this step is a set of program documents that define the methodologies to be employed across the organization, explaining how they embody the chosen frameworks and how they work together as a coherent system.
Step 3: The Document Pyramid as Implementation Structure
With frameworks translated into methodologies, we use the document pyramid to structure their implementation throughout the organization. This involves creating procedures, work instructions, and records that bring methodologies to life in day-to-day operations.
Key aspects of this step include:
- Procedure Development: At the Procedure level, we define who does what, when, and in what sequence. Procedures establish the process flows that implement methodologies without specifying detailed steps.
- Work Instruction Creation: At the Work Instruction level, we provide detailed guidance on how to perform specific tasks. Work instructions translate methodological steps into practical actions.
- Record Definition: At the Records level, we establish what evidence will be collected to demonstrate that processes are working as intended. Records provide feedback for evaluation and improvement.
The document pyramid ensures that there’s a clear line of sight from high-level frameworks to day-to-day activities, with each level providing appropriate detail for its intended audience and purpose.
Step 4: Tool Selection Criteria Derived from Higher Levels
With the structure in place, we can now establish criteria for tool selection that ensure alignment with frameworks and methodologies. These criteria are derived from the higher levels of the document pyramid, ensuring that tool selection serves overall system objectives.
Key criteria for tool selection include:
- Framework Alignment: Does the tool embody the principles of our chosen frameworks? Tools should reinforce rather than contradict the conceptual foundation of the quality system.
- Methodological Fit: Does the tool support the systematic approach defined in our methodologies? Tools should be appropriate for the specific methodology they’re implementing.
- System Integration: Does the tool integrate with other tools and systems? Tools should contribute to overall system coherence rather than creating silos.
- User Needs: Does the tool address the needs and capabilities of its users? Tools should be accessible and valuable to the people who will use them.
- Value Contribution: Does the tool provide value that justifies its cost and complexity? Tools should deliver benefits that outweigh their implementation and maintenance costs.
These criteria ensure that tool selection is guided by frameworks and methodologies rather than by trends or personal preferences.
Step 5: Evaluating Tools Against Framework Principles
Finally, we evaluate specific tools against our selection criteria and the principles of good systems design. This evaluation ensures that the tools we choose not only fulfill specific functions but also contribute to the coherence and effectiveness of the overall quality system.
For each tool under consideration, we ask:
- Balance: Does this tool address the needs of multiple stakeholders, or does it serve only limited interests?
- Congruence: Is this tool aligned with our frameworks, methodologies, and other tools?
- Convenience: Is this tool user-friendly and practical for regular use?
- Coordination: Does this tool work harmoniously with other components of our system?
- Elegance: Does this tool provide sufficient functionality without unnecessary complexity?
- Human-Centered: Does this tool enhance rather than diminish the human experience?
- Learning: Does this tool provide opportunities for reflection and improvement?
- Sustainability: Will this tool provide lasting value, or will it quickly become obsolete?
Tools that score well across these dimensions are more likely to contribute to a coherent and effective quality system than those that excel in only one or two areas.
The result of this structured approach is a deliberate path from frameworks to tools that ensures coherence, effectiveness, and sustainability in the quality system. Each tool is selected not in isolation but as part of a coherent whole, guided by frameworks and methodologies that provide context and direction.
Maturity Models: Tracking Implementation Progress
As organizations implement the framework-to-tool path, they need ways to assess their progress and identify areas for improvement. Maturity models provide structured frameworks for this assessment, helping organizations benchmark their current state and plan their development journey.
Understanding Maturity Models as Assessment Frameworks
Maturity models are structured frameworks used to assess the effectiveness, efficiency, and adaptability of an organization’s processes. They provide a systematic methodology for evaluating current capabilities and guiding continuous improvement efforts.
Key characteristics of maturity models include:
- Assessment and Classification: Maturity models help organizations understand their current process maturity level and identify areas for improvement.
- Guiding Principles: These models emphasize a process-centric approach focused on continuous improvement, aligning improvements with business goals, standardization, measurement, stakeholder involvement, documentation, training, technology enablement, and governance.
- Incremental Levels: Maturity models typically define a progression through distinct levels, each building on the capabilities of previous levels.
The Business Process Maturity Model (BPMM)
The Business Process Maturity Model is a structured framework for assessing and improving the maturity of an organization’s business processes. It provides a systematic methodology to evaluate the effectiveness, efficiency, and adaptability of processes within an organization, guiding continuous improvement efforts.
The BPMM typically consists of five incremental levels, each building on the previous one:
Initial Level: Ad-hoc Tool Selection
At this level, tool selection is chaotic and unplanned. Organizations exhibit these characteristics:
- Tools are selected arbitrarily without connection to frameworks or methodologies
- Different departments use different tools for similar purposes
- There’s limited understanding of the relationship between frameworks, methodologies, and tools
- Documentation is inconsistent and often incomplete
- The “magpie syndrome” is in full effect, with tools collected based on current trends or personal preferences
Managed Level: Consistent but Localized Selection
At this level, some structure emerges, but it remains limited in scope:
- Basic processes for tool selection are established but may not fully align with organizational frameworks
- Some risk assessment is used in tool selection, but not consistently
- Subject matter experts are involved in selection, but their roles are unclear
- There’s increased awareness of the need for justification in tool selection
- Tools may be selected consistently within departments but vary across the organization
Standardized Level: Organization-wide Approach
At this level, a consistent approach to tool selection is implemented across the organization:
- Tool selection processes are standardized and align with organizational frameworks
- Risk-based approaches are consistently used to determine tool requirements and priorities
- Subject matter experts are systematically involved in the selection process
- The concept of the framework-to-tool path is understood and applied
- The document pyramid is used to structure implementation
- Quality management principles guide tool selection criteria
Predictable Level: Data-Driven Tool Selection
At this level, quantitative measures are used to guide and evaluate tool selection:
- Key Performance Indicators (KPIs) for tool effectiveness are established and regularly monitored
- Data-driven decision-making is used to continually improve tool selection processes
- Advanced risk management techniques predict and mitigate potential issues with tool implementation
- There’s a strong focus on leveraging supplier documentation and expertise to streamline tool selection
- Engineering procedures for quality activities are formalized and consistently applied
- Return on investment calculations guide tool selection decisions
Optimizing Level: Continuous Improvement in Selection Process
At the highest level, the organization continuously refines its approach to tool selection:
- There’s a culture of continuous improvement in tool selection processes
- Innovation in selection approaches is encouraged while maintaining alignment with frameworks
- The organization actively contributes to developing industry best practices in tool selection
- Tool selection activities are seamlessly integrated with other quality management systems
- Advanced technologies may be leveraged to enhance selection strategies
- The organization regularly reassesses its frameworks and methodologies, adjusting tool selection accordingly
Applying Maturity Models to Tool Selection Processes
To effectively apply these maturity models to the framework-to-tool path, organizations should:
- Assess Current State: Evaluate your current tool selection practices against the maturity model levels. Identify your organization’s position on each dimension.
- Identify Gaps: Determine the gap between your current state and desired future state. Prioritize areas for improvement based on strategic objectives and available resources.
- Develop Improvement Plan: Create a roadmap for advancing to higher maturity levels. Define specific actions, responsibilities, and timelines.
- Implement Changes: Execute the improvement plan, monitoring progress and adjusting as needed.
- Reassess Regularly: Periodically reassess maturity levels to track progress and identify new improvement opportunities.
By using maturity models to guide the evolution of their framework-to-tool path, organizations can move systematically from ad-hoc tool selection to a mature, deliberate approach that ensures coherence and effectiveness in their quality systems.
Practical Implementation Strategy
Translating the framework-to-tool path from theory to practice requires a structured implementation strategy. This section outlines a practical approach for organizations at any stage of maturity, from those just beginning their journey to those refining mature systems.
Assessing Current State of Tool Selection Practices
Before implementing changes, organizations must understand their current approach to tool selection. This assessment should examine:
Documentation Structure: Does your organization have a defined document pyramid? Are there clear policies, programs, procedures, work instructions, and records?
Framework Clarity: Have you explicitly defined the frameworks that guide your quality efforts? Are these frameworks documented and understood by key stakeholders?
Selection Processes: How are tools currently selected? Who makes these decisions, and what criteria do they use?
Coherence Evaluation: To what extent do your current tools work together as a coherent system rather than a collection of individual instruments?
Maturity Level: Sssess your organization’s current maturity in tool selection practices.
This assessment provides a baseline from which to measure progress and identify priority areas for improvement. It should involve stakeholders from across the organization to ensure a comprehensive understanding of current practices.
Identifying Framework Gaps and Misalignments
With a clear understanding of current state, the next step is to identify gaps and misalignments in your framework-to-tool path:
Framework Definition Gaps: Are there areas where frameworks are undefined or unclear? Do stakeholders have a shared understanding of guiding principles?
Translation Breaks: Are frameworks effectively translated into methodologies through program-level documents? Is there a clear connection between high-level principles and operational approaches?
Procedure Inconsistencies: Do procedures align with defined methodologies? Do they provide clear guidance on who, what, and when without overspecifying how?
Tool-Framework Misalignments: Do current tools align with and support organizational frameworks? Are there tools that contradict or undermine framework principles?
Document Hierarchy Gaps: Are there missing or inconsistent elements in your document pyramid? Are connections between levels clearly established?
These gaps and misalignments highlight areas where the framework-to-tool path needs strengthening. They become the focus of your implementation strategy.
Documenting the Selection Process Through the Document Pyramid
With gaps identified, the next step is to document a structured approach to tool selection using the document pyramid:
Policy Level: Develop policy documents that clearly articulate your chosen frameworks and their guiding principles. These documents should establish the “what” of your quality system without specifying the “how”.
Program Level: Create program documents that translate frameworks into methodologies. These documents should serve as connective tissue, showing how frameworks are implemented through systematic approaches.
Procedure Level: Establish procedures for tool selection that define roles, responsibilities, and process flow. These procedures should outline who is involved in selection decisions, what criteria they use, and when these decisions occur.
Work Instruction Level: Develop detailed work instructions for tool evaluation and implementation. These should provide step-by-step guidance for assessing tools against selection criteria and implementing them effectively.
Records Level: Define the records to be maintained throughout the tool selection process. These provide evidence that the process is being followed and create a knowledge base for future decisions.
This documentation creates a structured framework-to-tool path that guides all future tool selection decisions.
Creating Tool Selection Criteria Based on Framework Principles
With the process documented, the next step is to develop specific criteria for evaluating potential tools:
Framework Alignment: How well does the tool embody and support your chosen frameworks? Does it contradict any framework principles?
Methodological Fit: Is the tool appropriate for your defined methodologies? Does it support the systematic approaches outlined in your program documents?
Systems Principles Application: How does the tool perform against the eight principles of good systems (Balance, Congruence, Convenience, Coordination, Elegance, Human-Centered, Learning, Sustainability)?
Integration Capability: How well does the tool integrate with existing systems and other tools? Does it contribute to system coherence or create silos?
User Experience: Is the tool accessible and valuable to its intended users? Does it enhance rather than complicate their work?
Value Proposition: Does the tool provide value that justifies its cost and complexity? What specific benefits does it deliver, and how do these align with organizational objectives?
These criteria should be documented in your procedures and work instructions, providing a consistent framework for evaluating all potential tools.
Implementing Review Processes for Tool Efficacy
Once tools are selected and implemented, ongoing review ensures they continue to deliver value and remain aligned with frameworks:
Regular Assessments: Establish a schedule for reviewing existing tools against framework principles and selection criteria. This might occur annually or when significant changes in context occur.
Performance Metrics: Define and track metrics that measure each tool’s effectiveness and contribution to system objectives. These metrics should align with the specific value proposition identified during selection.
User Feedback Mechanisms: Create channels for users to provide feedback on tool effectiveness and usability. This feedback is invaluable for identifying improvement opportunities.
Improvement Planning: Develop processes for addressing identified issues, whether through tool modifications, additional training, or tool replacement.
These review processes ensure that the framework-to-tool path remains effective over time, adapting to changing needs and contexts.
Tracking Maturity Development Using Appropriate Models
Finally, organizations should track their progress in implementing the framework-to-tool path using maturity models:
Maturity Assessment: Regularly assess your organization’s maturity using the BPMM, PEMM, or similar models. Document current levels across all dimensions.
Gap Analysis: Identify gaps between current and desired maturity levels. Prioritize these gaps based on strategic importance and feasibility.
Improvement Roadmap: Develop a roadmap for advancing to higher maturity levels. This roadmap should include specific initiatives, timelines, and responsibilities.
Progress Tracking: Monitor implementation of the roadmap, tracking progress toward higher maturity levels. Adjust strategies as needed based on results and changing circumstances.
By systematically tracking maturity development, organizations can ensure continuous improvement in their framework-to-tool path, gradually moving from ad-hoc selection to a fully optimized approach.
This practical implementation strategy provides a structured approach to establishing and refining the framework-to-tool path. By following these steps, organizations at any maturity level can improve the coherence and effectiveness of their tool selection processes.
Common Pitfalls and How to Avoid Them
While implementing the framework-to-tool path, organizations often encounter several common pitfalls that can undermine their efforts. Understanding these challenges and how to address them is essential for successful implementation.
The Technology-First Trap
Pitfall: One of the most common errors is selecting tools based on technological appeal rather than alignment with frameworks and methodologies. This “technology-first” approach is the essence of the magpie syndrome, where organizations are attracted to shiny new tools without considering their fit within the broader system.
Signs you’ve fallen into this trap:
- Tools are selected primarily based on features and capabilities
- Framework and methodology considerations come after tool selection
- Selection decisions are driven by technical teams without broader input
- New tools are implemented because they’re trendy, not because they address specific needs
How to avoid it:
- Always start with frameworks and methodologies, not tools
- Establish clear selection criteria based on framework principles
- Involve diverse stakeholders in selection decisions, not just technical experts
- Require explicit alignment with frameworks for all tool selections
- Use the five key questions of system design to evaluate any new technology
Ignoring the Human Element in Tool Selection
Pitfall: Tools are ultimately used by people, yet many organizations neglect the human element in selection decisions. Tools that are technically powerful but difficult to use or that undermine human capabilities often fail to deliver expected benefits.
Signs you’ve fallen into this trap:
- User experience is considered secondary to technical capabilities
- Training and change management are afterthoughts
- Tools require extensive workarounds in practice
- Users develop “shadow systems” to circumvent official tools
- High resistance to adoption despite technical superiority
How to avoid it:
- Include users in the selection process from the beginning
- Evaluate tools against the “Human” principle of good systems
- Consider the full user journey, not just isolated tasks
- Prioritize adoption and usability alongside technical capabilities
- Be empathetic with users, understanding their situation and feelings
- Implement appropriate training and support mechanisms
- Balance standardization with flexibility to accommodate user needs
Inconsistency Between Framework and Tools
Pitfall: Even when organizations start with frameworks, they often select tools that contradict framework principles or undermine methodological approaches. This inconsistency creates confusion and reduces effectiveness.
Signs you’ve fallen into this trap:
- Tools enforce processes that conflict with stated methodologies
- Multiple tools implement different approaches to the same task
- Framework principles are not reflected in daily operations
- Disconnection between policy statements and operational reality
- Confusion among staff about “the right way” to approach tasks
How to avoid it:
- Explicitly map tool capabilities to framework principles during selection
- Use the program level of the document pyramid to ensure proper translation from frameworks to tools
- Create clear traceability from frameworks to methodologies to tools
- Regularly audit tools for alignment with frameworks
- Address inconsistencies promptly through reconfiguration, replacement, or reconciliation
- Ensure selection criteria prioritize framework alignment
Misalignment Between Different System Levels
Pitfall: Without proper coordination, different levels of the quality system can become misaligned. Policies may say one thing, procedures another, and tools may enforce yet a third approach.
Signs you’ve fallen into this trap:
- Procedures don’t reflect policy requirements
- Tools enforce processes different from documented procedures
- Records don’t provide evidence of policy compliance
- Different departments interpret frameworks differently
- Audit findings frequently identify inconsistencies between levels
How to avoid it:
- Use the enhanced document pyramid to create clear connections between levels
- Ensure each level properly translates requirements from the level above
- Review all system levels together when making changes
- Establish governance mechanisms that ensure alignment
- Create visual mappings that show relationships between levels
- Implement regular cross-level reviews
- Use the “Congruence” and “Coordination” principles to evaluate alignment
Lack of Documentation and Institutional Memory
Pitfall: Many organizations fail to document their framework-to-tool path adequately, leading to loss of institutional memory when key personnel leave. Without documentation, decisions seem arbitrary and inconsistent over time.
Signs you’ve fallen into this trap:
- Selection decisions are not documented with clear rationales
- Framework principles exist but are not formally recorded
- Tool implementations vary based on who led the project
- Tribal knowledge dominates over documented processes
- New staff struggle to understand the logic behind existing systems
How to avoid it:
- Document all elements of the framework-to-tool path in the document pyramid
- Record selection decisions with explicit rationales
- Create and maintain framework and methodology documentation
- Establish knowledge management practices for preserving insights
- Use the “Learning” principle to build reflection and documentation into processes
- Implement succession planning for key roles
- Create orientation materials that explain frameworks and their relationship to tools
Failure to Adapt: The Static System Problem
Pitfall: Some organizations successfully implement a framework-to-tool path but then treat it as static, failing to adapt to changing contexts and requirements. This rigidity eventually leads to irrelevance and bypassing of formal systems.
Signs you’ve fallen into this trap:
- Frameworks haven’t been revisited in years despite changing context
- Tools are maintained long after they’ve become obsolete
- Increasing use of “exceptions” and workarounds
- Growing gap between formal processes and actual work
- Resistance to new approaches because “that’s not how we do things”
How to avoid it:
- Schedule regular reviews of frameworks and methodologies
- Use the “Learning” and “Sustainability” principles to build adaptation into systems2
- Establish processes for evaluating and incorporating new approaches
- Monitor external developments in frameworks, methodologies, and tools
- Create feedback mechanisms that capture changing needs
- Develop change management capabilities for system evolution
- Use maturity models to guide continuous improvement
By recognizing and addressing these common pitfalls, organizations can increase the effectiveness of their framework-to-tool path implementation. The key is maintaining vigilance against these tendencies and establishing practices that reinforce the principles of good system design.
Case Studies: Success Through Deliberate Selection
To illustrate the practical application of the framework-to-tool path, let’s examine three case studies from different industries. These examples demonstrate how organizations have successfully implemented deliberate tool selection guided by frameworks, with measurable benefits to their quality systems.
Case Study 1: Pharmaceutical Manufacturing Quality System Redesign
Organization: A mid-sized pharmaceutical manufacturer facing increasing regulatory scrutiny and operational inefficiencies.
Initial Situation: The company had accumulated dozens of quality tools over the years, with minimal coordination between them. Documentation was extensive but inconsistent, and staff complained about “check-box compliance” that added little value. Different departments used different approaches to similar problems, and there was no clear alignment between high-level quality objectives and daily operations.
Framework-to-Tool Path Implementation:
- Framework Selection: The organization adopted a dual framework approach combining ICH Q10 (Pharmaceutical Quality System) with Systems Thinking principles. These frameworks were documented in updated quality policies that emphasized a holistic approach to quality.
- Methodology Translation: At the program level, they developed a Quality System Master Plan that translated these frameworks into specific methodologies, including risk-based decision-making, knowledge management, and continuous improvement. This document served as connective tissue between frameworks and operational procedures.
- Procedure Development: Procedures were redesigned to align with the selected methodologies, clearly defining roles, responsibilities, and processes. These procedures emphasized what needed to be done and by whom without overspecifying how tasks should be performed.
- Tool Selection: Tools were evaluated against criteria derived from the frameworks and methodologies. This evaluation led to the elimination of redundant tools, reconfiguration of others, and the addition of new tools where gaps existed. Each tool was documented in work instructions that connected it to higher-level requirements.
- Maturity Tracking: The organization used PEMM to assess their initial maturity and track progress over time, developing a roadmap for advancing from P-2 (basic standardization) to P-4 (optimization).
Results: Two years after implementation, the organization achieved:
- 30% decrease in deviation investigations through improved root cause analysis
- Successful regulatory inspections with zero findings
- Improved staff engagement in quality activities
- Advancement from P-2 to P-3 on the PEMM maturity scale
Key Lessons:
- The program-level documentation was crucial for translating frameworks into operational practices
- The deliberate evaluation of tools against framework principles eliminated many inefficiencies
- Maturity modeling provided a structured approach to continuous improvement
- Executive sponsorship and cross-functional involvement were essential for success
Case Study 2: Medical Device Design Transfer Process
Organization: A growing medical device company struggling with inconsistent design transfer from R&D to manufacturing.
Initial Situation: The design transfer process involved multiple departments using different tools and approaches, resulting in delays, quality issues, and frequent rework. Teams had independently selected tools based on familiarity rather than appropriateness, creating communication barriers and inconsistent outputs.
Framework-to-Tool Path Implementation:
- Framework Selection: The organization adopted the Quality by Design (QbD) framework integrated with Design Controls requirements from 21 CFR 820.30. These frameworks were documented in a new Design Transfer Policy that established principles for knowledge-based transfer.
- Methodology Translation: A Design Transfer Program document was created to translate these frameworks into methodologies, specifically Stage-Gate processes, Risk-Based Design Transfer, and Knowledge Management methodologies. This document mapped how different approaches would work together across the product lifecycle.
- Procedure Development: Cross-functional procedures defined responsibilities across departments and established standardized transfer points with clear entrance and exit criteria. These procedures created alignment without dictating specific technical approaches.
- Tool Selection: Tools were evaluated against framework principles and methodological requirements. This led to standardization on a core set of tools, including Design Failure Mode Effects Analysis (DFMEA), Process Failure Mode Effects Analysis (PFMEA), Design of Experiments (DoE), and Statistical Process Control (SPC). Each tool was documented with clear connections to higher-level requirements.
- Maturity Tracking: The organization used BPMM to assess and track their maturity in the design transfer process, initially identifying themselves at Level 2 (Managed) with a goal of reaching Level 4 (Predictable).
Results: 18 months after implementation, the organization achieved:
- 50% reduction in design transfer cycle time
- 60% reduction in manufacturing defects related to design transfer issues
- Improved first-time-right performance in initial production runs
- Better cross-functional collaboration and communication
- Advancement from Level 2 to Level 3+ on the BPMM scale
Key Lessons:
- The QbD framework provided a powerful foundation for selecting appropriate tools
- Standardizing on a core toolset improved cross-functional communication
- The program document was essential for creating a coherent approach
- Regular maturity assessments helped maintain momentum for improvement
Lessons Learned from Successful Implementations
Across these diverse case studies, several common factors emerge as critical for successful implementation of the framework-to-tool path:
- Executive Sponsorship: In all cases, senior leadership commitment was essential for establishing frameworks and providing resources for implementation.
- Cross-Functional Involvement: Successful implementations involved stakeholders from multiple departments to ensure comprehensive perspective and buy-in.
- Program-Level Documentation: The program level of the document pyramid consistently proved crucial for translating frameworks into operational approaches.
- Deliberate Tool Evaluation: Taking the time to systematically evaluate tools against framework principles and methodological requirements led to more coherent and effective toolsets.
- Maturity Modeling: Using maturity models to assess current state, set targets, and track progress provided structure and momentum for continuous improvement.
- Balanced Standardization: Successful implementations balanced the need for standardization with appropriate flexibility for different contexts.
- Clear Documentation: Comprehensive documentation of the framework-to-tool path created transparency and institutional memory.
- Continuous Assessment: Regular evaluation of tool effectiveness against framework principles ensured ongoing alignment and adaptation.
These lessons provide valuable guidance for organizations embarking on their own journey from frameworks to tools. By following these principles and adapting them to their specific context, organizations can achieve similar benefits in quality, efficiency, and effectiveness.
Summary of Key Principles
Several fundamental principles emerge as essential for establishing an effective framework-to-tool path:
- Start with Frameworks: Begin with the conceptual foundations that provide structure and guidance for your quality system. Frameworks establish the “what” and “why” before addressing the “how”.
- Use the Document Pyramid: The enhanced document pyramid – with policies, programs, procedures, work instructions, and records – provides a coherent structure for implementing your framework-to-tool path.
- Apply Systems Thinking: The eight principles of good systems (Balance, Congruence, Convenience, Coordination, Elegance, Human-Centered, Learning, Sustainability) serve as evaluation criteria throughout the journey.
- Build Coherence: True coherence goes beyond alignment, creating systems that build order through their function rather than through rigid control.
- Think Before Implementing: Understand system purpose, structure, behavior, and context – rather than simply implementing technology.
- Follow a Structured Approach: The five-step approach (Framework Selection → Methodology Translation → Document Pyramid Implementation → Tool Selection Criteria → Tool Evaluation) provides a systematic path from concepts to implementation.
- Track Maturity: Maturity models help assess current state and guide continuous improvement in your framework-to-tool path.
These principles provide a foundation for transforming tool selection from a haphazard collection of shiny objects to a deliberate implementation of coherent strategy.
The Value of Deliberate Selection in Professional Practice
The deliberate selection of tools based on frameworks offers numerous benefits over the “magpie” approach:
Coherence: Tools work together as an integrated system rather than a collection of disconnected parts.
Effectiveness: Tools directly support strategic objectives and methodological approaches.
Efficiency: Redundancies are eliminated, and resources are focused on tools that provide the greatest value.
Sustainability: The system adapts and evolves while maintaining its essential character and purpose.
Engagement: Staff understand the “why” behind tools, increasing buy-in and proper utilization.
Learning: The system incorporates feedback and continuously improves based on experience.
These benefits translate into tangible outcomes: better quality, lower costs, improved regulatory compliance, enhanced customer satisfaction, and increased organizational capability.
Next Steps for Implementing in Your Organization
If you’re ready to implement the framework-to-tool path in your organization, consider these practical next steps:
- Assess Current State: Evaluate your current approach to tool selection using the maturity models described earlier. Identify your organization’s maturity level and key areas for improvement.
- Document Existing Frameworks: Identify and document the frameworks that currently guide your quality efforts, whether explicit or implicit. These form the foundation for your path.
- Enhance Your Document Pyramid: Review your documentation structure to ensure it includes all necessary levels, particularly the crucial program level that connects frameworks to operational practices.
- Develop Selection Criteria: Based on your frameworks and the principles of good systems, create explicit criteria for tool selection and document these criteria in your procedures.
- Evaluate Current Tools: Assess your existing toolset against these criteria, identifying gaps, redundancies, and misalignments. Based on this evaluation, develop an improvement plan.
- Create a Maturity Roadmap: Develop a roadmap for advancing your organization’s maturity in tool selection. Define specific initiatives, timelines, and responsibilities.
- Implement and Monitor: Execute your improvement plan, tracking progress against your maturity roadmap. Adjust strategies based on results and changing circumstances.
These steps will help you establish a deliberate path from frameworks to tools that enhances the coherence and effectiveness of your quality system.
The journey from frameworks to tools represents a fundamental shift from the “magpie syndrome” of haphazard tool collection to a deliberate approach that creates coherent, effective quality systems. Organizations can transform their tool selection processes by following the principles and techniques outlined here and significantly improve quality, efficiency, and effectiveness. The document pyramid provides the structure, maturity models track the progress, and systems thinking principles guide the journey. The result is better tool selection and a truly integrated quality system that delivers sustainable value.



.jpg){kind=link}