Flowing Data Does it again with a great and helpful primer. A definite must read.
Author: Jeremiah Genest
Quality professional with over 20 years of experience. Gamer and storyteller with forty years of practice.
The Role of the HACCP
Reading Strukmyer LLC’s recent FDA Warning Letter, and reflecting back to last year’s Colgate-Palmolive/Tom’s of Maine, Inc. Warning Letter, has me thinking of common language In both warning letters where the FDA asks for “A comprehensive, independent assessment of the design and control of your firm’s manufacturing operations, with a detailed and thorough review of all microbiological hazards.”
It is hard to read that as anything else than a clarion call to use a HACCP.
If that isn’t a HACCP, I don’t know what is. Given the FDA’s rich history and connection to the tool, it is difficult to imagine them thinking of any other tool. Sure, I can invent about 7 other ways to do that, but why bother when there is a great tool, full of powerful uses, waiting to be used that the regulators pretty much have in their DNA.

The Evolution of HACCP in FDA Regulation: A Journey to Enhanced Food Safety
The Hazard Analysis and Critical Control Points (HACCP) system has a fascinating history that is deeply intertwined with FDA regulations. Initially developed in the 1960s by NASA, the Pillsbury Company, and the U.S. Army, HACCP was designed to ensure safe food for space missions. This pioneering collaboration aimed to prevent food safety issues by identifying and controlling critical points in food processing. The success of HACCP in space missions soon led to its application in commercial food production.
In the 1970s, Pillsbury applied HACCP to its commercial operations, driven by incidents such as the contamination of farina with glass. This prompted Pillsbury to adopt HACCP more widely across its production lines. A significant event in 1971 was a panel discussion at the National Conference on Food Protection, which led to the FDA’s involvement in promoting HACCP for food safety inspections. The FDA recognized the potential of HACCP to enhance food safety standards and began to integrate it into its regulatory framework.
As HACCP gained prominence as a food safety standard in the 1980s and 1990s, the National Advisory Committee on Microbiological Criteria for Foods (NACMCF) refined its principles. The committee added preliminary steps and solidified the seven core principles of HACCP, which include hazard analysis, critical control points identification, establishing critical limits, monitoring procedures, corrective actions, verification procedures, and record-keeping. This structured approach helped standardize HACCP implementation across different sectors of the food industry.
A major milestone in the history of HACCP was the implementation of the Pathogen Reduction/HACCP Systems rule by the USDA’s Food Safety and Inspection Service (FSIS) in 1996. This rule mandated HACCP in meat and poultry processing facilities, marking a significant shift towards preventive food safety measures. By the late 1990s, HACCP became a requirement for all food businesses, with some exceptions for smaller operations. This widespread adoption underscored the importance of proactive food safety management.
The Food Safety Modernization Act (FSMA) of 2011 further emphasized preventive controls, including HACCP, to enhance food safety across the industry. FSMA shifted the focus from responding to food safety issues to preventing them, aligning with the core principles of HACCP. Today, HACCP remains a cornerstone of food safety management globally, with ongoing training and certification programs available to ensure compliance with evolving regulations. The FDA continues to support HACCP as part of its broader efforts to protect public health through safe food production and processing practices. As the food industry continues to evolve, the principles of HACCP remain essential for maintaining high standards of food safety and quality.
Why is a HACCP Useful in Biotech Manufacturing
The HACCP seeks to map a process – the manufacturing process, one cleanroom, a series of interlinked cleanrooms, or the water system – and identifies hazards (a point of contamination) by understanding the personnel, material, waste, and other parts of the operational flow. These hazards are assessed at each step in the process for their likelihood and severity. Mitigations are taken to reduce the risk the hazard presents (“a contamination control point”). Where a risk cannot be adequately minimized (either in terms of its likelihood of occurrence, the severity of its nature, or both), this “contamination control point” should be subject to a form of detection so that the facility has an understanding of whether the microbial hazard was potentially present at a given time, for a given operation. In other words, the “critical control point” provides a reasoned area for selecting a monitoring location. For aseptic processing, for example, the target is elimination, even if this cannot be absolutely demonstrated.
The HACCP approach can easily be applied to pharmaceutical manufacturing where it proves very useful for microbial control. Although alternative risk tools exist, such as Failure Modes and Effects Analysis, the HACCP approach is better for microbial control.
The HACCP is a core part of an effective layers of control analysis.
Conducting a HACCP

HACCP provides a systematic approach to identifying and controlling potential hazards throughout the production process.
Step 1: Conduct a Hazard Analysis
- List All Process Steps: Begin by detailing every step involved in your biotech manufacturing process, from raw material sourcing to final product packaging. Make sure to walk down the process thoroughly.
- Identify Potential Hazards: At each step, identify potential biological, chemical, and physical hazards. Biological hazards might include microbial contamination, while chemical hazards could involve chemical impurities or inappropriate reagents. Physical hazards might include particulates or inappropriate packaging materials.
- Evaluate Severity and Likelihood: Assess the severity and likelihood of each identified hazard. This evaluation helps prioritize which hazards require immediate attention.
- Determine Preventive Measures: Develop strategies to control significant hazards. This might involve adjusting process conditions, improving cleaning protocols, or enhancing monitoring systems.
- Document Justifications: Record the rationale behind including or excluding hazards from your analysis. This documentation is essential for transparency and regulatory compliance.
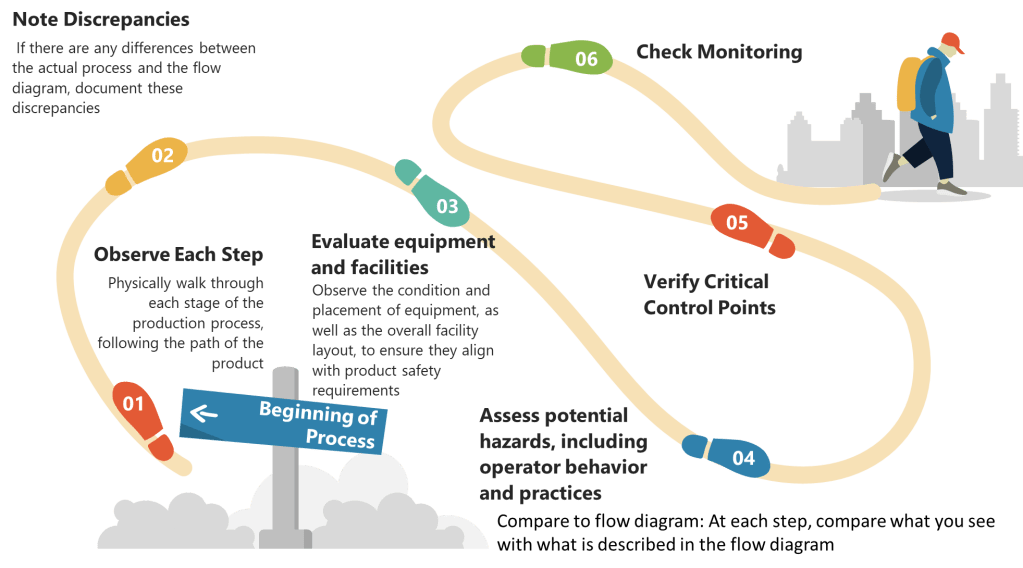
Step 2: Determine Critical Control Points (CCPs)
- Identify Control Points: Any step where biological, chemical, or physical factors can be controlled is considered a control point.
- Determine CCPs: Use a decision tree to identify which control points are critical. A CCP is a step at which control can be applied and is essential to prevent or eliminate a hazard or reduce it to an acceptable level.
- Establish Critical Limits: For each CCP, define the maximum or minimum values to which parameters must be controlled. These limits ensure that hazards are effectively managed.
| Control Points | Critical Control Points |
| Process steps where a control measure (mitigation activity) is necessary to prevent the hazard from occurring | Process steps where both control and monitoring are necessary to assure product quality and patient safety |
| Are not necessarily critical control points (CCPs) | Are also control points |
| Determined from the risk associated with the hazard | Determined through a decision tree |
Step 3: Establish Monitoring Procedures
- Develop Monitoring Plans: Create detailed plans for monitoring each CCP. This includes specifying what to monitor, how often, and who is responsible.
- Implement Monitoring Tools: Use appropriate tools and equipment to monitor CCPs effectively. This might include temperature sensors, microbial testing kits, or chemical analyzers.
- Record Monitoring Data: Ensure that all monitoring data is accurately recorded and stored for future reference.
Step 4: Establish Corrective Actions
- Define Corrective Actions: Develop procedures for when monitoring indicates that a CCP is not within its critical limits. These actions should restore control and prevent hazards.
- Proceduralize: You are establishing alternative control strategies here so make sure they are appropriately verified and controlled by process/procedure in the quality system.
- Train Staff: Ensure that all personnel understand and can implement corrective actions promptly.
Step 5: Establish Verification Procedures
- Regular Audits: Conduct regular audits to verify that the HACCP system is functioning correctly. This includes reviewing monitoring data and observing process operations.
- Validation Studies: Perform validation studies to confirm that CCPs are effective in controlling hazards.
- Continuous Improvement: Use audit findings to improve the HACCP system over time.
Step 6: Establish Documentation and Record-Keeping
- Maintain Detailed Records: Keep comprehensive records of all aspects of the HACCP system, including hazard analyses, CCPs, monitoring data, corrective actions, and verification activities.
- Ensure Traceability: Use documentation to ensure traceability throughout the production process, facilitating quick responses to any safety issues.
Step 7: Implement and Review the HACCP Plan
- Implement the Plan: Ensure that all personnel involved in biotech manufacturing understand and follow the HACCP plan.
- Regular Review: Regularly review and update the HACCP plan to reflect changes in processes, new hazards, or lessons learned from audits and incidents.

Worker’s Empowerment
Empowerment is a foundational element of a quality culture, where workers are entrusted with the authority to make decisions, initiate actions, and take responsibility for the outcomes of their work. This approach not only enhances job satisfaction and productivity but also fosters a culture of autonomy and participation, which is essential for achieving high organizational performance. However, the concept of empowerment has sometimes been misinterpreted within quality management frameworks such as Total Quality Management (TQM), Lean, and Six Sigma. In these contexts, empowerment rhetoric is occasionally used to justify increased work demands and managerial oversight, rather than genuinely empowering workers to contribute to quality improvements. A true quality culture, therefore, requires a genuine commitment to empowering workers, ensuring that they have the autonomy to drive continuous improvement and innovation.
History of Worker Empowerment
The concept of empowerment has its roots in social movements, including the civil rights and women’s rights movements, where it was used to describe the process of gaining autonomy and self-determination for marginalized groups. In the context of management, empowerment gained prominence in the 1980s and 1990s as a way to improve organizational performance by engaging workers more effectively.
Several management thinkers have discussed and advocated for worker empowerment, contributing significantly to the development of this concept. Here are some key figures and their contributions:
Mary Parker Follett
- Autonomy and Collective Power: Follett emphasized the importance of giving workers autonomy to complete their jobs effectively. She believed that when workers have the freedom to work independently, they become happier, more productive, and more engaged. Follett’s “power with” principle suggests that power should be shared among many, rather than concentrated in a few hands, fostering a collaborative environment.
- Collaboration and Flexibility: Follett advocated for establishing personal ownership of company goals while allowing flexibility in achieving them. This approach encourages agile problem-solving and creative solutions that benefit the business.
Tom Peters
- Self-Managing Teams: Peters has been a strong advocate for creating self-managing teams where leadership roles rotate among members. He emphasizes the importance of listening to workers and believing in their unlimited potential. Peters’ philosophy includes empowering front-line staff to act as business teams, which can significantly enhance organizational performance.
- Empowerment through Leadership: Peters suggests that managers should be retrained to become listeners rather than talkers, fostering an environment where every worker feels valued and empowered to contribute.
W. Edwards Deming
- Involvement and Autonomy: Deming’s 14 Points for Management include principles that support worker empowerment, such as removing barriers to pride of workmanship and encouraging collaboration across departments. These principles aim to create an environment where workers feel valued and empowered to improve processes.
- Continuous Improvement: Deming’s emphasis on continuous improvement processes, like kaizen, involves worker participation, which can be seen as a form of empowerment. However, it is crucial to ensure that such participation is genuine and not merely rhetorical.
Rosabeth Moss Kanter
- Change Management: Kanter’s change management theory emphasizes creating a collaborative and transparent work environment. Her approach involves empowering worker by encouraging them to speak up, team up, and continuously work towards positive change within the organization.
- Empowerment through Participation: Kanter’s principles promote worker engagement and loyalty by involving them in organizational changes and decision-making processes.
Elton Mayo
- Human Relations Theory: Mayo’s work highlights the importance of social and relational factors in motivating workers. While not directly focused on empowerment, his theory suggests that workers are more motivated by attention and camaraderie than by monetary rewards alone. This perspective supports the idea that empowering workers involves recognizing their social needs and fostering a supportive work environment.
These thinkers have contributed to the understanding and implementation of worker empowerment by emphasizing autonomy, collaboration, and the importance of recognizing employee contributions. Their ideas continue to influence management practices today.
Dimensions of Empowerment
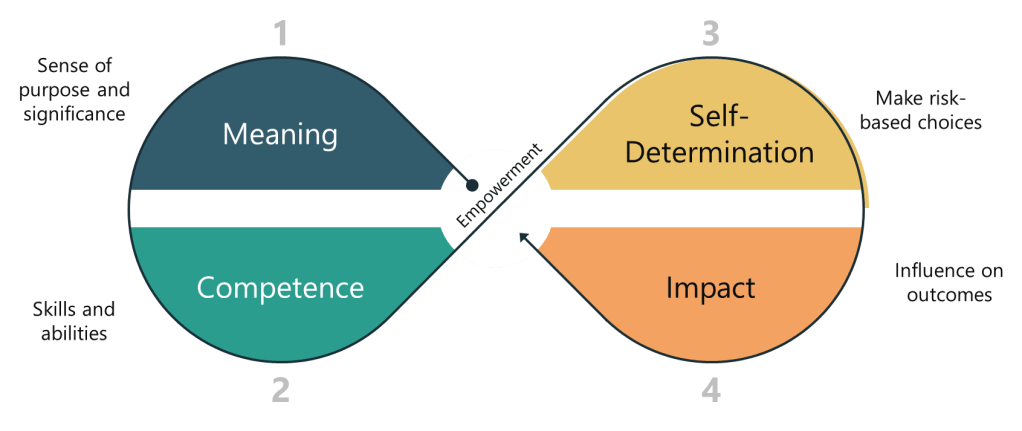
Empowerment can be understood through several key dimensions:
- Meaning: This refers to the sense of purpose and significance that employees derive from their work. When employees feel that their work is meaningful, they are more likely to be motivated and engaged.
- Competence: This dimension involves the skills and abilities that employees need to perform their jobs effectively. Empowerment requires that employees have the necessary competencies to make decisions and take actions.
- Self-Determination: This is the ability of employees to make choices and decisions about their work. Self-determination is crucial for empowerment, as it allows employees to feel in control of their tasks and outcomes.
- Impact: This dimension refers to the influence that employees have on organizational outcomes. When employees feel that their actions can make a difference, they are more likely to be empowered and motivated.
Implementation Practices
Implementing empowerment effectively requires several key practices:
- Clear Communication: Employees need clear expectations and goals to understand how their work contributes to the organization’s objectives.
- Training and Development: Providing employees with the necessary skills and knowledge to make informed decisions is essential for empowerment.
- Autonomy and Decision-Making Authority: Employees should have the freedom to make decisions within their scope of work.
- Feedback and Recognition: Regular feedback and recognition of employee contributions help reinforce empowerment by acknowledging their impact.
Deming’s Involvement in Worker Empowerment
W. Edwards Deming, a pioneer in quality management, emphasized the importance of employee involvement and empowerment through his 14 Points for Management. Specifically:
- Point 3: Cease dependence on inspection to achieve quality. Eliminate the need for inspection on a mass basis by building quality into the product in the first place. This point encourages organizations to empower workers by giving them the tools and training needed to ensure quality during production.
- Point 9: Break down barriers between departments. People in research, design, sales, and production must work as a team to foresee problems of production and in use that may be encountered with the product or service. This emphasizes collaboration and cross-functional teamwork, which is a form of empowerment.
- Point 12: Remove barriers that rob the hourly worker of his right to pride of workmanship. The responsibility of supervisors must be changed from sheer numbers to quality. This point directly addresses the need to empower workers by removing obstacles that prevent them from taking pride in their work.
Deming’s philosophy aligns with genuine empowerment by focusing on building quality into processes, fostering teamwork, and recognizing the value of worker pride and autonomy.
Denison and Organizational Culture
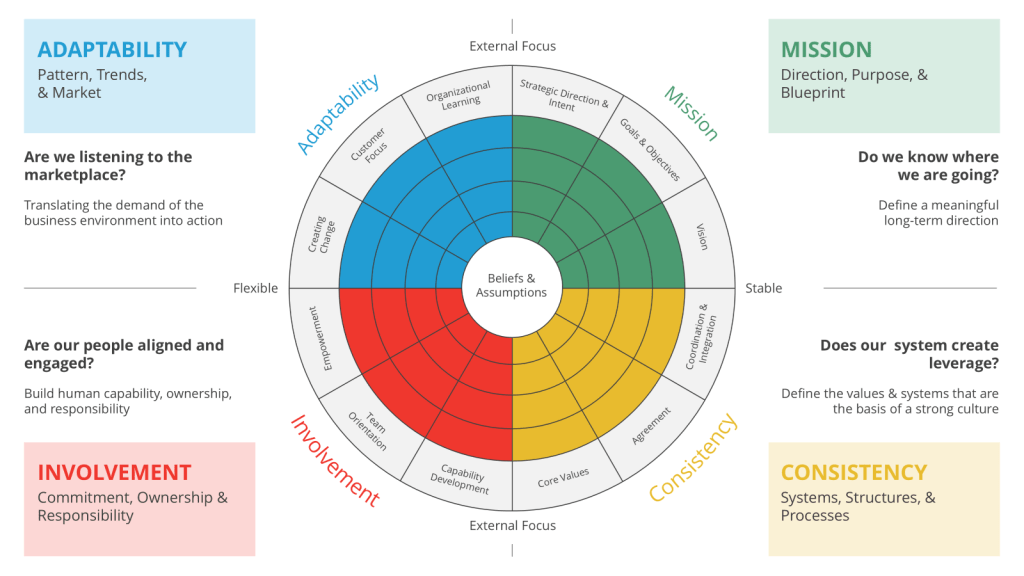
Daniel Denison’s work on organizational culture, particularly through the Denison Model, assesses culture across four critical traits: Mission, Involvement, Adaptability, and Consistency. Each of these traits is further divided into three indexes, providing a comprehensive framework for understanding and improving organizational culture.
Involvement and Empowerment
Denison’s model emphasizes the importance of Involvement, which is the degree to which individuals at all levels are engaged and feel a sense of ownership in the organization. This trait is crucial for empowerment, as it involves aligning employees with the business direction and positioning them to contribute to its success. The indexes under Involvement include aspects such as empowerment, team orientation, and capability development, all of which are essential for creating a culture where employees feel valued and empowered.
Empowerment through Cultural Alignment
Denison suggests that empowerment is not just about giving employees authority but also about ensuring they are aligned with and committed to the organization’s mission. By fostering a culture where workers are engaged and capable, organizations can enhance their performance metrics such as innovation, customer satisfaction, and worker satisfaction. Denison’s approach emphasizes the need for leaders to manage culture effectively, recognizing that culture can either support or hinder organizational goals.
Leadership and Empowerment
Denison’s model implies that leaders should focus on creating an environment where workers feel empowered to contribute. This involves not only setting a clear mission but also ensuring that systems and processes support worker involvement and adaptability. By doing so, leaders can foster a culture where workers are motivated to drive organizational success. Denison’s philosophy underscores the importance of balancing internal consistency with external adaptability, ensuring that organizations remain responsive to market changes while maintaining internal cohesion.
Denison’s work provides a structured framework for understanding how empowerment fits into a broader organizational culture. By emphasizing involvement and alignment, organizations can create an environment where workers feel empowered to contribute to success.
Misuse of Empowerment Rhetoric in Quality Methodologies
Total Quality Management (TQM)
TQM emphasizes worker involvement and empowerment as part of its comprehensive approach to quality improvement. However, the emphasis on continuous improvement and customer satisfaction can sometimes lead to increased workloads and stress for workers, undermining genuine empowerment.
Lean Manufacturing
Lean manufacturing focuses on eliminating waste and maximizing efficiency, often using empowerment rhetoric to encourage workers to participate in continuous improvement processes like kaizen. However, this can result in workers being manipulated into accepting intensified workloads without real control over their conditions.
Six Sigma
Six Sigma uses a structured approach to quality improvement, relying on trained professionals like Green and Black Belts. While it involves worker participation, the focus on defect reduction and process optimization can lead to a narrow definition of empowerment that serves managerial goals rather than worker autonomy.
Avoiding the Misuse of Empowerment Rhetoric
To avoid misusing empowerment rhetoric, organizations should focus on creating a genuine culture of empowerment by:
Ensuring Autonomy
Ensuring autonomy in the workplace is crucial for empowering workers. This involves providing them with real decision-making authority and the freedom to act within their roles. When workers have autonomy, they are more likely to feel a sense of ownership over their work, which can lead to increased motivation and productivity. Autonomy allows workers to make decisions that align with their expertise and judgment, reducing the need for constant managerial oversight. This not only speeds up decision-making processes but also fosters a culture of trust and responsibility. To implement autonomy effectively, organizations should clearly define the scope of decision-making authority for each role, ensure that workers understand their responsibilities, and provide the necessary resources and support to facilitate independent action. By doing so, organizations can create an environment where workers feel valued and empowered to contribute to organizational success.
Fostering Meaningful Work
Fostering meaningful work is essential for creating a sense of purpose and engagement among workers. This involves aligning worker tasks with organizational goals and ensuring that work contributes to a broader sense of purpose. When workers understand how their tasks fit into the larger picture, they are more likely to be motivated and committed to their work. Meaningful work encourages workers to see beyond their immediate tasks and understand the impact of their contributions on the organization and its stakeholders. To foster meaningful work, organizations should communicate clearly about organizational objectives and how individual roles contribute to these goals. Additionally, providing opportunities for workers to participate in goal-setting and strategic planning can enhance their sense of purpose and connection to the organization’s mission. By making work meaningful, organizations can create a workforce that is not only productive but also passionate about achieving shared objectives.
Developing Competence
Developing competence is a critical aspect of empowering workers . This involves investing in training and development to enhance their skills and abilities. When workers feel competent in their roles, they are more confident and capable of making decisions and taking initiatives. Competence development should be tailored to the needs of both the organization and the individual worker, ensuring that training programs are relevant and effective. Organizations should also provide ongoing opportunities for learning and growth, recognizing that competence is not static but rather something that evolves over time. By investing in worker development, organizations can create a skilled and adaptable workforce that is better equipped to handle challenges and drive innovation. Moreover, when workers see that their employer is committed to their growth, they are more likely to feel valued and committed to the organization.
Recognizing Impact
Recognizing the impact of workers contributions is vital for reinforcing their sense of empowerment. Regularly acknowledging and rewarding worker achievements helps to demonstrate that their work is valued and appreciated. This can be done through various means, such as public recognition, bonuses, or promotions. However, recognition should be genuine and specific, highlighting the specific contributions and outcomes that workers have achieved. Generic or superficial recognition can undermine its effectiveness and lead to skepticism among workers. To make recognition meaningful, organizations should establish clear criteria for what constitutes impactful work and ensure that recognition is timely and consistent. By acknowledging workers contributions, organizations can foster a culture of appreciation and motivation, encouraging workers to continue striving for excellence and making significant contributions to organizational success.
Encouraging Self-Determination
Encouraging self-determination is essential for empowering workers to take ownership of their work processes and outcomes. This involves supporting workers in making choices about how they complete their tasks and achieve their objectives. Self-determination allows workers to work in ways that best suit their skills and work styles, leading to increased job satisfaction and productivity. To encourage self-determination, organizations should provide workers with the flexibility to design their work processes and set their own goals, as long as these align with organizational objectives. Additionally, organizations should foster an environment where workers feel comfortable suggesting improvements and innovations, without fear of criticism or reprisal. By giving workers the autonomy to make decisions about their work, organizations can tap into their creativity and initiative, leading to more effective and efficient work processes. This approach not only empowers workers but also contributes to a more agile and responsive organization.
By focusing on these aspects, organizations can move beyond rhetorical empowerment and create a truly empowered workforce.
Conclusion
Worker empowerment is a powerful concept that, when implemented genuinely, can lead to significant improvements in organizational performance and worker satisfaction. However, its misuse in quality methodologies like TQM, Lean, and Six Sigma can undermine its potential benefits. By understanding the dimensions of empowerment and aligning practices with Deming’s principles, organizations can foster a culture of true empowerment that benefits both workers and the organization as a whole.
Building a Safe Space for Reflection: Leveraging Psychological Safety Towards a Quality Culture
Creating a safe space for reflection is crucial for fostering innovation, problem-solving, and continuous improvement. This environment is deeply rooted in psychological safety and a quality culture, where employees feel empowered to express themselves freely, share ideas, and challenge existing norms without fear of judgment or reprisal.
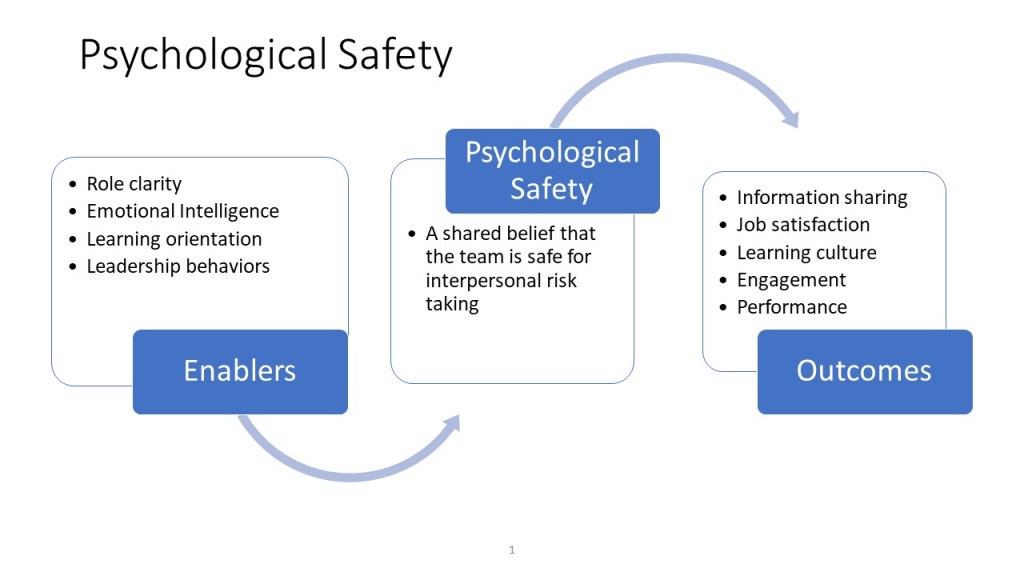
Understanding Psychological Safety
Psychological safety refers to a shared belief among team members that they are safe to take risks, share their thoughts, and learn from their mistakes without fear of negative consequences. This concept is foundational to building a culture where individuals feel valued, included, and motivated to contribute their unique perspectives. It is the bedrock upon which effective collaboration, creativity, and problem-solving are built. In environments where psychological safety is prioritized, employees are more likely to engage in open dialogue, admit mistakes, and explore new ideas, leading to enhanced innovation and productivity.
The Role of Leadership in Fostering Psychological Safety
Effective leadership plays a pivotal role in establishing and maintaining a culture of psychological safety. Leaders must set the tone by modeling vulnerability, encouraging open communication, and demonstrating empathy towards their team members. They should establish clear expectations of respect and inclusivity, ensuring that diverse perspectives are welcomed and valued. By doing so, leaders create an environment where employees feel comfortable sharing their thoughts and ideas, which is essential for driving innovation and solving complex problems.
In the past post on Psychological Safety, Reflexivity, and Problem Solving, I explored how psychological safety enables individuals to behave authentically and express themselves candidly, which is crucial for effective problem-solving and reflexivity in organizations. This authenticity allows teams to tackle challenges more effectively by leveraging diverse viewpoints and experiences.
Building a Quality Culture
A quality culture is deeply intertwined with psychological safety. It emphasizes continuous improvement, learning from mistakes, and a commitment to excellence. In such a culture, employees are encouraged to reflect on their processes, identify areas for improvement, and implement changes that enhance overall performance. This reflective practice is facilitated by psychological safety, as it allows individuals to share insights and ideas without fear of criticism, thereby fostering a collaborative and adaptive environment.
Strategies for Creating a Safe Space for Reflection
Creating a safe space for reflection involves several strategic steps:
Establishing Open Communication Channels
Organizations should implement transparent and constructive communication channels that allow employees to express their thoughts, concerns, and ideas without fear of negative consequences. This can be achieved through regular team meetings, anonymous feedback systems, or open forums where employees feel comfortable sharing their perspectives. Active listening and empathy are crucial in these interactions, as they reinforce the sense of safety and encourage further participation.
Implementing Psychological Safety Training
Providing comprehensive training on psychological safety is essential for building awareness and equipping employees with the skills needed to navigate complex interactions and support their colleagues. These programs should emphasize the importance of trust, vulnerability, and inclusivity, and offer practical strategies for fostering a psychologically safe environment. By educating employees on these principles, organizations can ensure that psychological safety becomes an integral part of their culture.
Encouraging Active Participation and Feedback
Encouraging active participation involves creating opportunities for employees to engage in collaborative discussions and provide feedback. This can be facilitated through workshops, brainstorming sessions, or project meetings where diverse perspectives are sought and valued. Feedback loops should be open and constructive, allowing employees to learn from their experiences and grow professionally.
Measuring Psychological Safety
Measuring psychological safety is critical for understanding its impact on organizational culture and identifying areas for improvement. This can be achieved through surveys, behavioral indicators, and engagement scores. Surveys should include questions that assess employees’ perceptions of safety, trust, and openness within their teams. Behavioral indicators, such as the frequency of idea sharing and openness in feedback loops, can also provide valuable insights into the level of psychological safety within an organization.
In our previous discussions on on this blog, I have emphasized the importance of a culture that supports open dialogue and continuous improvement. A few examples include:
- “Communication Loops and Silos: A Barrier to Effective Decision Making in Complex Industries“: This post highlights the challenges of communication loops and silos in industries like aviation and biotechnology. It emphasizes the need for open dialogue to bridge these gaps and improve decision-making processes.
- “Change Strategies for Accelerating Change“: This post discusses strategies such as promoting cross-functional training, fostering informal interactions, and implementing feedback loops. These strategies are crucial for creating a culture that supports open dialogue and continuous improvement.
- “Reducing Subjectivity in Quality Risk Management: Aligning with ICH Q9(R1)“: This post focuses on reducing subjectivity through structured approaches and data-driven decision-making. It underscores the importance of a culture that encourages open communication to ensure that decisions are based on comprehensive data rather than personal biases.
These examples illustrate the importance of fostering a culture that supports open dialogue and continuous improvement in complex industries.
Overcoming Challenges
Despite the benefits of psychological safety, several challenges may arise when attempting to implement it within an organization. Fear and resistance to change are common obstacles, particularly in hierarchical structures where speaking up can be perceived as risky. To overcome these challenges, organizations should identify influential champions who can model psychological safety behaviors and inspire others to do the same. Regular assessments and feedback sessions can also help identify areas where psychological safety is lacking, allowing for targeted interventions.
Sustaining Psychological Safety
Sustaining a culture of psychological safety requires ongoing effort and commitment. Organizations must regularly assess the effectiveness of their psychological safety initiatives and refine their strategies based on feedback and performance data. This involves ensuring that leadership behaviors consistently reinforce psychological safety principles and that training programs are scaled to reach all levels of the organization.
Conclusion
Building a safe space for reflection within an organization is a multifaceted process that relies heavily on psychological safety and a quality culture. By fostering an environment where employees feel valued, included, and empowered to share their ideas, organizations can unlock their full potential and drive innovation. Psychological safety is not a static state but a continuous journey that requires leadership commitment, effective communication, and ongoing evaluation. As we continue to navigate the complexities of modern organizational challenges, prioritizing psychological safety will remain essential for creating a workplace where employees thrive and contribute meaningfully.
By embracing psychological safety and fostering a quality culture, organizations can create a safe space for reflection that drives innovation, enhances collaboration, and promotes continuous improvement. This approach not only benefits the organization but also contributes to the well-being and growth of its employees, ultimately leading to a more resilient and adaptive workforce.
Safecor Health Warning Letter Closeout
I got a post on my RSS feed today from the FDA for a closeout letter to Safecor Health in response to the 2023 Warning Letter. Always happy to see an actual closeout letter.
The main takeaways from the FDA warning letter:
Inadequate Line Clearance and Packaging Controls:
- Safecor failed to properly inspect packaging and labeling facilities before use, leading to potential mix-ups of drug products. This was evidenced by the presence of unrelated tablets and capsules during the packaging of a specific product.
- The company has a history of product mix-ups, including instances where a vitamin was found in a drug meant to prevent organ transplant rejection and mislabeled blood clot prevention medication.
Insufficient Cleaning and Maintenance Procedures:
- The firm lacked adequate procedures for cleaning and maintaining equipment, with unidentified residues found on supposedly clean equipment. This poses a risk of cross-contamination.
- The company’s cleaning validation program was deemed inadequate, particularly in addressing worst-case scenarios.
Failure to Test Components:
- Safecor did not adequately test incoming components, such as water used in drug manufacturing, for purity, strength, and quality.
- The company relied on visual inspections without performing necessary chemical and microbiological tests.
Quality Control Unit Deficiencies:
- The quality control unit failed to ensure compliance with CGMP regulations, including inadequate document control and data integrity issues.
- Manufacturing records were not properly controlled, and corrections were made using correction fluid, raising concerns about data authenticity.
