Engineering runs (ERs) represent a critical yet often underappreciated component of modern biopharmaceutical validation strategies. Defined as non-GMP-scale trials that simulate production processes to identify risks and optimize parameters, Engineering Runs bridge the gap between theoretical process design and manufacturing. Their integration into the ASTM E2500 verification framework creates a powerful synergy – combining Good Engineering Practice (GEP) with Quality Risk Management (QRM) to meet evolving regulatory expectations.
When aligned with ICH Q10’s pharmaceutical quality system (PQS) and the ASTM E2500 lifecycle approach, ERs transform from operational exercises into strategic tools for:
Design space verification per ICH Q8
Scale-up risk mitigation during technology transfer
Specification & Design The standard mandates “right-sized” documentation – detailed enough to ensure product quality without unnecessary bureaucracy.
Verification This phase provides a unified verification approach focusing on:
Critical process parameters (CPPs)
Worst-case scenario testing
Leveraging vendor testing data
Acceptance & Release Final review incorporates ICH Q10’s management responsibilities, ensuring traceability from initial risk assessments to verification outcomes.
Engineering runs serve as a critical bridge between design verification and formal Process Performance Qualification (PPQ). ERs validate critical aspects of manufacturing systems by confirming:
Equipment functionality under simulated GMP conditions
Process parameter boundaries for Critical Process Parameters (CPPs)
Facility readiness through stress-testing utilities, workflows, and contamination controls
Demonstration/ Training Run prior to GMP area
Shakedown. Demonstration/Training Run in GMP area
Engineering Run
cGMP Manufacturing
Room and Equipment
Room
N/A
IOQ Post-Approval
Released and Active
Process Gas
Generation and Distribution Released Point of use assembly PQ complete
Process utility
Process Equipment
Functionally verified or calibrated as required (commissioned)
IOQ Approved
Full released
Analytical Equipment
Released
Alarms
N/A
Alarm ranges and plan defined
Alarms qualified
Raw Materials
Bill of Materials
RM in progress
Approved
Suppliers
Approval in Progress
Approved
Specifications
In Draft
Effective
Release
Non-GMP Usage decision
Released
Process Documentation
Source Documentation
To be defined in Tech Transfer Plan
Engineering Run Protocol
Tech Transfer closed
Batch Records and product specific Work Instructions
Draft
Reviewed Draft
Approved
Process and Equipment SOPs
N/A
Draft
Effective
Product Labels
N/A
Draft Labels
Approved Labels
QC Testing and Documentation
BSC and Personnel Environmental Monitoring
N/A
Effective
Analytical Methods
Suitable for use
Phase Appropriate Validation
Stability
N/A
In place
Certificate of Analysis
N/A
Defined in Engineering Protocol
Effective
Sampling Plan
Draft
Draft use as defined in engineering protocol
Effective
Operations/Execution
Operator Training
Observe and perform operations to gain hands on experience with SME observation
Process specific equipment OJT Gown qualifiedBSC OJT Aseptic OJT Material Transfer OJT (All training in eQMS)
Training in Use
Process Lock
As defined in Tech Transfer Plan
6-week prior to execution
Approved Process Description
Deviations
N/A
N/A
Process – Per Engineering Run protocol FUSE – per SOP
The pharmaceutical industry is navigating a transformative period in contamination control, driven by the convergence of updated international standards. The U.S. Pharmacopeia’s draft chapter〈1110〉 Microbial Contamination Control Strategy Considerations (March 2025) joins EU GMP Annex 1 (2022) in emphasizing risk-based strategies but differ in technical requirements and classification systems.
USP〈1110〉: A Lifecycle-Oriented Microbial Control Framework
The draft USP chapter introduces a comprehensive contamination control strategy (CCS) that spans the entire product lifecycle, from facility design to post-market surveillance. It emphasizes microbial, endotoxin, and pyrogen risks, requiring manufacturers to integrate quality risk management (QRM) into every operational phase. Facilities must adopt ISO 14644-1 cleanroom classifications, with ISO Class 5 (≤3,520 particles ≥0.5 µm/m³) mandated for aseptic processing areas. Environmental monitoring programs must include both viable (microbial) and nonviable particles, with data trends analyzed quarterly to refine alert/action levels. Unlike Annex 1, USP allows flexibility in risk assessment methodologies but mandates documented justifications for control measures, such as the use of closed systems or isolators to minimize human intervention.
EU GMP Annex 1: Granular Cleanroom and Sterilization Requirements
Annex 1 builds on ISO 14644-1 cleanroom standards but introduces pharmaceutical-specific adaptations through its Grade A–D system. Grade A zones (critical processing areas) require ISO Class 5 conditions during both “at-rest” and “in-operation” states, with continuous particle monitoring and microbial limits of <1 CFU/m³. Annex 1 also mandates smoke studies to validate unidirectional airflow patterns in Grade A areas, a requirement absent in ISO 14644-1. Sterilization processes, such as autoclaving and vaporized hydrogen peroxide (VHP) treatments, require pre- and post-use integrity testing, aligning with its focus on sterility assurance.
Reconciling Annex 1 and ISO 14644-1 Cleanroom Classifications
While both frameworks reference ISO 14644-1, Annex 1 overlays additional pharmaceutical requirements:
Aspect
EU GMP Annex 1
ISO 14644-1
Classification System
Grades A–D mapped to ISO classes
ISO Class 1–9 based on particle counts
Particle Size
≥0.5 µm and ≥5.0 µm monitoring for Grades A–B
≥0.1 µm to ≥5.0 µm, depending on class
Microbial Limits
Explicit CFU/m³ limits for each grade
No microbial criteria; focuses on particles
Operational States
Qualification required for “at-rest” and “in-operation” states
Single-state classification permitted
Airflow Validation
Smoke studies mandatory for Grade A
Airflow pattern testing optional
For example, a Grade B cleanroom (ISO Class 7 at rest) must maintain ISO Class 7 particle counts during production but adheres to stricter microbial limits (≤10 CFU/m³) than ISO 14644-1 alone. Manufacturers must design monitoring programs that satisfy both standards, such as deploying continuous particle counters for Annex 1 compliance while maintaining ISO certification reports.
Classification
Description
Grade A
Critical area for high-risk and aseptic operations that corresponds to ISO 5 at rest/static and ISO 4.8 (in-operation/dynamic). Grade A areas apply to aseptic operations where the sterile product, product primary packaging components and product-contact surfaces are exposed to the environment. Normally Grade A conditions are provided by localized air flow protection, such as unidirectional airflow workstations within a Restricted Access Barrier System (RABS) or isolator. Direct intervention (e.g., without the protection of barrier and glove port protection) into the Grade A area by operators must be minimized by premises, equipment, process, or procedural design.
Grade B
For aseptic preparation and filling, this is the background area for Grade A (where it is not an isolator) and corresponds to ISO 5 at rest/static and ISO 7 in-operation/dynamic. Air pressure differences must be continuously monitored. Classified spaces of lower grade can be considered with the appropriate risk assessment and technical justification.
Grade C
Used for carrying out less critical steps in the manufacture of aseptically filled sterile products or as a background for isolators. They can also be used for the preparation/filling of terminally sterilized products. Grade C correspond to ISO 7 at rest/static and ISO 8 in-operation/dynamic.
Grade D
Used to carry out non-sterile operations and corresponds to ISO 8 at rest/static and in-operation/dynamic.
Both frameworks require Quality Risk Management. USP〈1110〉advocates for a flexible, science-driven approach, allowing tools like HACCP (Hazard Analysis Critical Control Points) or FMEA (Failure Modes Effects Analysis) to identify critical control points. For instance, a biologics manufacturer might use HACCP to prioritize endotoxin controls during cell culture harvesting. USP also emphasizes lifecycle risk reviews, requiring CCS updates after facility modifications or adverse trend detections.
Annex 1 mandates formal QRM processes with documented risk assessments for all sterilization and aseptic processes. Its Annex 1.25 clause requires FMEA for media fill simulations, ensuring worst-case scenarios (e.g., maximum personnel presence) are tested. Risk assessments must also justify cleanroom recovery times after interventions, linking airflow validation data to contamination probability.
A harmonized approach involves:
Baseline Risk Identification: Use HACCP to map contamination risks across product stages, aligning with USP’s lifecycle focus.
Control Measure Integration: Apply Annex 1’s sterilization and airflow requirements to critical risks identified in USP’s CCS.
Continuous Monitoring: Combine USP’s trend analysis with continuous monitoring for real-time risk mitigation.
Strategic Implementation Considerations
Reconciling these standards requires a multi-layered strategy. Facilities must first achieve ISO 14644-1 certification for particle counts, then overlay Annex 1’s microbial and operational requirements. For example, an ISO Class 7 cleanroom used for vial filling would need Grade B microbial monitoring (≤10 CFU/m³) and quarterly smoke studies to validate airflow. Risk management documentation should cross-reference USP’s CCS objectives with Annex 1’s sterilization validations, creating a unified audit trail. Training programs must blend USP’s aseptic technique modules with Annex 1’s cleanroom behavior protocols, ensuring personnel understand both particle control and microbial hygiene.
Toward Global Harmonization
The draft USP〈1110〉and Annex 1 represent complementary pillars of modern contamination control. By anchoring cleanroom designs to ISO 14644-1 and layering region-specific requirements, manufacturers can streamline compliance across jurisdictions. Proactive risk management—combining USP’s flexibility with Annex 1’s rigor—will be pivotal in navigating this evolving landscape. As regulatory expectations converge, firms that invest in integrated CCS platforms will gain agility in an increasingly complex global market.
What the Warning Letter Reveals About Process Validation
The FDA’s inspection identified several violations that directly pertain to inadequate process validation. Process validation is essential for ensuring that drug manufacturing processes consistently produce products meeting their intended specifications. Here are the notable findings:
Failure to Validate Sterilization Processes:
The firm did not establish adequate controls to prevent microbiological contamination in drug products purporting to be sterile. Specifically, it relied on sterilization processes without monitoring pre-sterilization bioburden or maintaining appropriate environmental conditions.
The FDA emphasized that sterility testing alone is insufficient to assure product safety. It must be part of a broader validation strategy that includes pre-sterilization controls and environmental monitoring.
Inadequate Validation of Controlled-Release Dosage Forms:
The company failed to demonstrate that its controlled-release products conformed to specifications for active ingredient release rates. This lack of validation raises concerns about therapeutic efficacy and patient safety.
The response provided by the firm was deemed inadequate as it lacked retrospective assessments of marketed products and a detailed plan for corrective actions.
Insufficient Procedures for Production and Process Control:
The firm increased batch sizes without validating the impact on product quality and failed to include critical process parameters in batch records.
The FDA highlighted the importance of process qualification studies, which evaluate intra-batch variations and establish a state of control before commercial distribution.
Key Learnings for Pharmaceutical Manufacturers
The violations outlined in this warning letter provide valuable lessons for manufacturers aiming to maintain CGMP compliance:
Comprehensive Process Validation is Non-Negotiable
Process validation must encompass all stages of manufacturing, from raw materials to finished products. Manufacturers should:
Conduct rigorous qualification studies before scaling up production.
Validate sterilization processes, including pre-sterilization bioburden testing, environmental controls, and monitoring systems.
Sterility Testing Alone is Insufficient
Sterility testing should complement other preventive measures rather than serve as the sole assurance mechanism. Manufacturers must implement controls throughout the production lifecycle to minimize contamination risks.
Quality Control Units Must Exercise Oversight
The role of quality control units (QU) is pivotal in ensuring compliance across all operations, including oversight of contract testing laboratories and contract manufacturing organizations (CMOs). Failure to enforce proper testing protocols can lead to regulatory action.
Repeat Violations Signal Systemic Failures
The letter noted repeated violations from prior inspections in 2019 and 2021, indicating insufficient executive management oversight.
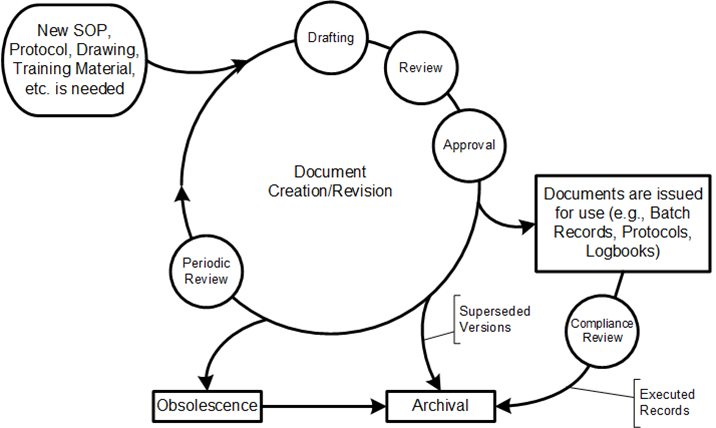
Review and document updates as needed (including reapprovals)
Managing changes and revision status
Ensuring availability of current versions
Maintaining document legibility and identification
Controlling distribution of external documents
This lifecycle usually has three critical dates associated with approval:
Approval Date: When designated authorities have reviewed and approved the document
Issuance Date: When the document is released into the document management system
Effective Date: When the document officially takes effect and must be followed
These dates are dependent on the type of document and can change as a result of workflow decisions.
Type of Document
Approval Date
Issuance date
Effective Date
Functional
Date Approved by final approver (sequential or parallel)
Date Training Made Available
End of Training Period
Record
Date Approved by final approver (sequential or parallel)
Usually automated to be same as Date Approved
Usually same as Date Approved
Report
Date Approved by final approver (sequential or parallel)
Usually automated to be same as Date Approved
Usually same as Date Approved
At the heart of the difference between these three days is the question of implementation and the Effective Date. At its core, the effective date is the date on which the requirements, instructions, or obligations in a document become binding for all affected parties. In the context of GxP document management, this represents the moment when:
Previous versions of the document are officially superseded
All operations must follow the new procedures outlined in the document
Training on the new procedures must be completed
Compliance audits will use the new document as their reference standard
One of the most frequently overlooked aspects of document management is the implementation period between document approval and its effective date. This period serves a critical purpose: ensuring that all affected personnel understand the document’s content and can execute its requirements correctly before it becomes binding.
In order to implement a new process change in a compliant manner, people must be trained in the new procedure before the document becomes effective. This fundamental principle ensures that by the time a new process goes “live,” everyone is prepared to perform the revised activity correctly and training records have been completed. Without this preparation period, organizations risk introducing non-compliance at the very moment they attempt to improve quality.
The implementation period bridges the gap between formal approval and practical application, addressing the human element of quality systems that automated solutions alone cannot solve.
Selecting Appropriate Implementation Periods
When configuring document change control systems, organizations must establish clear guidelines for determining implementation periods. The most effective approach is to build this determination into the change control workflow itself.
Several factors should influence the selection of implementation periods:
Urgency: In cases of immediate risk to patient safety or product quality, implementation periods may be compressed while still ensuring adequate training.
Risk Assessment: Higher-risk changes typically require more extensive training and therefore longer implementation periods.
Operational Impact: Changes affecting critical operations may need carefully staged implementation.
Training Complexity: Documents requiring hands-on training necessitate longer periods than read-only procedures.
Resource Availability: Consider the availability of trainers and affected personnel
Determining Appropriate Training Periods
The time required for training should be determined during the impact assessment phase of the change approval process. This assessment should consider:
The number of people requiring training
The complexity of the procedural changes
The type of training required (read-only versus observed assessment)
Operational constraints (shift patterns, production schedules)
Many organizations standardize on a default period (typically two weeks), but the most effective approach tailors the implementation period to each document’s specific requirements. For critical processes with many stakeholders, longer periods may be necessary, while simple updates affecting few staff might require only minimal time.
Consider this scenario: Your facility operates two shifts with 70 people during the day and 30 at night. An updated SOP requires all operators to complete not just read-only training but also a one-hour classroom assessment. If manufacturing schedules permit only 10 operators per shift to attend training, you would need a minimum of 7 days before the document becomes effective. Without this calculated implementation period, every operator would instantly become non-compliant when the new procedure takes effect.
The distinction between a procedure’s approval date and its effective date serves a critical purpose. This gap allows for proper training and implementation before the procedure becomes binding. However, there are specific circumstances when personnel might appropriately use a procedure they’ve been trained on before its official effective date.
1. Urgent Safety or Quality Concerns
When there is an immediate risk to patient safety or product quality, the time between approval and effectiveness may be compressed. For these cases there should be a mechanism to move up the effective date.
In such cases, the organization should prioritize training and implementation while still maintaining proper documentation of the accelerated timeline.
2. During Implementation Period for Training Purposes
The implementation period itself is designed to allow for training and controlled introduction of the new procedure. During this time, a limited number of trained personnel may need to use the new procedure to:
Train others on the new requirements
Test the procedure in a controlled environment
Prepare systems and equipment for the full implementation
These are all tasks that should be captured in the change control.
3. For Qualification and Validation Activities
During qualification protocol execution, procedures that have been approved but are not yet effective may be used under controlled conditions to validate systems, equipment, or processes. These activities typically occur before full implementation and are carefully documented to demonstrate compliance. Again these are captured in the change control and appropriate validation plan.
In some regulatory contexts, such as IRB approvals in clinical research, there are provisions for “approval with conditions” where certain activities may proceed before all requirements are finalized2. While not directly analogous to procedure implementation, this demonstrates regulatory recognition of staged implementation approaches.
Required Controls When Using Pre-Effective Procedures
If an organization determines it necessary to use an approved but not yet effective procedure, the following controls should be in place:
Documented Risk Assessment: A risk assessment should be conducted and documented to justify the early use of the procedure, especially considering potential impacts on product quality, data integrity, or patient safety.
Authorization: Special authorization from management and quality assurance should be obtained and documented.
Verification of Training: Evidence must be available confirming that the individuals using the procedure have been properly trained and assessed on the new requirements.
What About Parallel Compliance with Current Effective Procedures?
In all cases, the currently effective procedure must still be followed until the new procedure’s effective date. However there are changes, usually as a result of process improvement, usually in knowledge work processes where it is possible to use parts of the new procedure. For example, the new version of the deviation procedure adds additional requirements for assessing the deviation, or a new risk management tool is rolled out. In these cases you can meet the new compliance path without violating the current compliance path. The organization should demonstrate how both compliance paths are being maintained.
In cases where the new compliance path does not contain the old, but instead offers a new pathway, it is critical to maintain one way of work-as-prescribed and the effective date is a solid line.
Organizations should remember that the implementation period exists to ensure a smooth, compliant transition between procedures. Any exception to this standard approach should be carefully considered, well-justified, and thoroughly documented to maintain GxP compliance and minimize regulatory risk.
As an American Pharmaceutical Quality professional who has worked in and with European colleagues for decades, I am used to hearing, “But the requirements in country X are different,” to which my response is always, “Prove it.”
EudraLex represents the cornerstone of Good Manufacturing Practice (GMP) regulations within the European Union, providing a comprehensive framework that ensures medicinal products meet stringent quality, safety, and efficacy standards. You will understand the fundamentals if you know and understand Eudralex volume 4. However, despite this unified approach, a few specific national differences exist in how a select few of these regulations are interpreted and implemented – mostly around Qualified Persons, GMP certifications, registrations and inspection types.
EudraLex: The European Union Pharmaceutical Regulatory Framework
EudraLex serves as the cornerstone of pharmaceutical regulation in the European Union, providing a structured approach to ensuring medicinal product quality, safety, and efficacy. The framework is divided into several volumes, with Volume 4 specifically addressing Good Manufacturing Practice (GMP) for both human and veterinary medicinal products. The legal foundation for these guidelines stems from Directive 2001/83/EC, which establishes the Community code for medicinal products for human use, and Directive 2001/82/EC for veterinary medicinal products.
Within this framework, manufacturing authorization is mandatory for all pharmaceutical manufacturers in the EU, whether their products are sold within or outside the Union. Two key directives establish the principles and guidelines for GMP: Directive 2003/94/EC for human medicinal products and Directive 91/412/EEC for veterinary products. These directives are interpreted and implemented through the detailed guidelines in the Guide to Good Manufacturing Practice.
Structure and Implementation of EU Pharmaceutical Regulation
The EU pharmaceutical regulatory framework operates on multiple levels. At the highest level, EU institutions establish the legal framework through regulations and directives. EU Law includes both Regulations, which have binding legal force in every Member State, and Directives, which lay down outcomes that must be achieved while allowing each Member State some flexibility in transposing them into national laws.
The European Medicines Agency (EMA) coordinates and harmonizes at the EU level, while national regulatory authorities inspect, license, and enforce compliance locally. This multilayered approach ensures consistent quality standards while accommodating certain national considerations.
For marketing authorization, medicinal products may follow several pathways:
Authorizing body
Procedure
Scientific Assessment
Territorial scope
European Commission
Centralized
European Medicines Agency (EMA)
EU
National authorities
Mutual Recognition, Decentralized, National
National competent authorities (with possible additional assessment by EMA in case of disagreement)
EU countries concerned
This structure reflects the balance between EU-wide harmonization and national regulatory oversight in pharmaceutical manufacturing and authorization.
National Variations in Pharmaceutical Manufacturing Requirements
Austria
Austria maintains one of the more stringent interpretations of EU directives regarding Qualified Person requirements. While the EU directive 2001/83/EC establishes general qualifications for QPs, individual member states have some flexibility in implementing these requirements, and Austria has taken a particularly literal approach.
Austria also maintains a national “QP” or “eligible QP” registry, which is not a universal practice across all EU member states. This registry provides an additional layer of regulatory oversight and transparency regarding individuals qualified to certify pharmaceutical batches for release.
Denmark
Denmark has really flexible GMP certification recognition, but beyond that no real differences from Eudralex volume 4.
France
The Exploitant Status
The most distinctive feature of the French pharmaceutical regulatory framework is the “Exploitant” status, which has no equivalent in EU regulations. This status represents a significant departure from the standard European model and creates additional requirements for companies wishing to market medicinal products in France.
Under the French Public Health Code, the Exploitant is defined as “the company or organization providing the exploitation of medicinal products”. Exploitation encompasses a broad range of activities including “wholesaling or free distribution, advertising, information, pharmacovigilance, batch tracking and, where necessary, batch recall as well as any corresponding storage operations”. This status is uniquely French, as the European legal framework only recognizes three distinct positions: the Marketing Authorization Holder (MAH), the manufacturer, and the distributor.
The Exploitant status is mandatory for all companies that intend to market medicinal products in France. This requirement applies regardless of whether the product has received a standard marketing authorization or an early access authorization (previously known as Temporary Use Authorization or ATU).
To obtain and maintain Exploitant status, a company must fulfill several requirements that go beyond standard EU regulations:
The company must obtain a pharmaceutical establishment license authorized by the French National Agency for the Safety of Medicines and Health Products (ANSM).
It must employ a qualified person called a Chief Pharmaceutical Officer (Pharmacien Responsable).
It must designate a local qualified person for Pharmacovigilance.
The Pharmacien Responsable: A Unique French Pharmaceutical Role
Another distinctive feature of the French health code is the requirement for a Pharmacien Responsable (Chief Pharmaceutical Officer or CPO), a role with broader responsibilities than the “Qualified Person” defined at the European level.
According to Article L.5124-2 of the French Public Health Code, “any company operating a pharmaceutical establishment engaged in activities such as purchasing, manufacturing, marketing, importing or exporting, and wholesale distribution of pharmaceutical products must be owned by a pharmacist or managed by a company which management or general direction includes a Pharmacien Responsable”. This appointment is mandatory and serves as a prerequisite for any administrative authorization request to operate a pharmaceutical establishment in France.
The Pharmacien Responsable holds significant responsibilities and personal liability, serving as “a guarantor of the quality of the medication and the safety of the patients”. The role is deeply rooted in French pharmaceutical tradition, deriving “directly from the pharmaceutical monopoly” and applying to all pharmaceutical companies in France regardless of their activities.
The Pharmacien Responsable “primarily organizes and oversees all pharmaceutical operations (manufacturing, advertising, information dissemination, batch monitoring and recalls) and ensures that transportation conditions guarantee the proper preservation, integrity, and safety of products”. They have authority over delegated pharmacists, approve their appointments, and must be consulted regarding their departure.
The corporate mandate of the Pharmacien Responsable varies depending on the legal structure of the company, but their placement within the organizational hierarchy must clearly demonstrate their authority and responsibility. This requirement for clear placement in the company’s organization chart, with explicit mention of hierarchical links and delegations, has no direct equivalent in standard EU pharmaceutical regulations.
Germany
While Germany has many distinctive elements—including the PZN identification system, the securPharm verification approach, specialized distribution regulations, and nuanced clinical trial oversight—the GMPs from Eudralex Volume 4 are the same.
Italy
Italy has implemented a highly structured inspection system with clearly defined categories that create a distinctive national approach to GMP oversight.
National Preventive Inspections
Activating new manufacturing plants for active substances
Activating new manufacturing departments or lines
Reactivating departments that have been suspended
Authorizing manufacturing or import of new active substances (particularly sterile or biological products)
National Follow-up Inspections to verify the GMP compliance of the corrective actions declared as implemented by the manufacturing plant in the follow-up phase of a previous inspection. This structured approach to verification creates a continuous improvement cycle within the Italian regulatory system.
Extraordinary or Control Inspections: These are conducted outside normal inspection programs when necessary for public health protection.
Spain
The differences in Spain are mostly on the way an organization is registered and has no impacts on GMP operations.
Regulatory Recognition and Mutual Agreements
EU member states have received specific recognition for their GMP inspection capabilities from international partners individually.