Over the past decades, as I’ve grown and now led quality organizations in biotechnology, I’ve encountered many thinkers who’ve shaped my approach to investigation and risk management. But few have fundamentally altered my perspective like Sidney Dekker. His work didn’t just add to my toolkit—it forced me to question some of my most basic assumptions about human error, system failure, and what it means to create genuinely effective quality systems.
Dekker’s challenge to move beyond “safety theater” toward authentic learning resonates deeply with my own frustrations about quality systems that look impressive on paper but fail when tested by real-world complexity.
Why Dekker Matters for Quality Leaders
Professor Sidney Dekker brings a unique combination of academic rigor and operational experience to safety science. As both a commercial airline pilot and the Director of the Safety Science Innovation Lab at Griffith University, he understands the gap between how work is supposed to happen and how it actually gets done. This dual perspective—practitioner and scholar—gives his critiques of traditional safety approaches unusual credibility.
But what initially drew me to Dekker’s work wasn’t his credentials. It was his ability to articulate something I’d been experiencing but couldn’t quite name: the growing disconnect between our increasingly sophisticated compliance systems and our actual ability to prevent quality problems. His concept of “drift into failure” provided a framework for understanding why organizations with excellent procedures and well-trained personnel still experience systemic breakdowns.
The “New View” Revolution
Dekker’s most fundamental contribution is what he calls the “new view” of human error—a complete reframing of how we understand system failures. Having spent years investigating deviations and CAPAs, I can attest to how transformative this shift in perspective can be.
The Traditional Approach I Used to Take:
Human error causes problems
People are unreliable; systems need protection from human variability
Solutions focus on better training, clearer procedures, more controls
Dekker’s New View That Changed My Practice:
Human error is a symptom of deeper systemic issues
People are the primary source of system reliability, not the threat to it
Variability and adaptation are what make complex systems work
This isn’t just academic theory—it has practical implications for every investigation I lead. When I encounter “operator error” in a deviation investigation, Dekker’s framework pushes me to ask different questions: What made this action reasonable to the operator at the time? What system conditions shaped their decision-making? How did our procedures and training actually perform under real-world conditions?
This shift aligns perfectly with the causal reasoning approaches I’ve been developing on this blog. Instead of stopping at “failure to follow procedure,” we dig into the specific mechanisms that drove the event—exactly what Dekker’s view demands.
Drift Into Failure: Why Good Organizations Go Bad
Perhaps Dekker’s most powerful concept for quality leaders is “drift into failure”—the idea that organizations gradually migrate toward disaster through seemingly rational local decisions. This isn’t sudden catastrophic failure; it’s incremental erosion of safety margins through competitive pressure, resource constraints, and normalized deviance.
I’ve seen this pattern repeatedly. For example, a cleaning validation program starts with robust protocols, but over time, small shortcuts accumulate: sampling points that are “difficult to access” get moved, hold times get shortened when production pressure increases, acceptance criteria get “clarified” in ways that gradually expand limits.
Each individual decision seems reasonable in isolation. But collectively, they represent drift—a gradual migration away from the original safety margins toward conditions that enable failure. The contamination events and data integrity issues that plague our industry often represent the endpoint of these drift processes, not sudden breakdowns in otherwise reliable systems.
Traditional root cause analysis seeks the single factor that “caused” an event, but complex system failures emerge from multiple interacting conditions. The take-the-best heuristic I’ve been exploring on this blog—focusing on the most causally powerful factor—builds directly on Dekker’s insight that we need to understand mechanisms, not hunt for someone to blame.
When I investigate a failure now, I’m not looking for THE root cause. I’m trying to understand how various factors combined to create conditions for failure. What pressures were operators experiencing? How did procedures perform under actual conditions? What information was available to decision-makers? What made their actions reasonable given their understanding of the situation?
This approach generates investigations that actually help prevent recurrence rather than just satisfying regulatory expectations for “complete” investigations.
Just Culture: Moving Beyond Blame
Dekker’s evolution of just culture thinking has been particularly influential in my leadership approach. His latest work moves beyond simple “blame-free” environments toward restorative justice principles—asking not “who broke the rule” but “who was hurt and how can we address underlying needs.”
This shift has practical implications for how I handle deviations and quality events. Instead of focusing on disciplinary action, I’m asking: What systemic conditions contributed to this outcome? What support do people need to succeed? How can we address the underlying vulnerabilities this event revealed?
This doesn’t mean eliminating accountability—it means creating accountability systems that actually improve performance rather than just satisfying our need to assign blame.
Safety Theater: The Problem with Compliance Performance
Dekker’s most recent work on “safety theater” hits particularly close to home in our regulated environment. He defines safety theater as the performance of compliance when under surveillance that retreats to actual work practices when supervision disappears.
I’ve watched organizations prepare for inspections by creating impressive documentation packages that bear little resemblance to how work actually gets done. Procedures get rewritten to sound more rigorous, training records get updated, and everyone rehearses the “right” answers for auditors. But once the inspection ends, work reverts to the adaptive practices that actually make operations function.
This theater emerges from our desire for perfect, controllable systems, but it paradoxically undermines genuine safety by creating inauthenticity. People learn to perform compliance rather than create genuine safety and quality outcomes.
The falsifiable quality systems I’ve been advocating on this blog represent one response to this problem—creating systems that can be tested and potentially proven wrong rather than just demonstrated as compliant.
Six Practical Takeaways for Quality Leaders
After years of applying Dekker’s insights in biotechnology manufacturing, here are the six most practical lessons for quality professionals:
1. Treat “Human Error” as the Beginning of Investigation, Not the End
When investigations conclude with “human error,” they’ve barely started. This should prompt deeper questions: Why did this action make sense? What system conditions shaped this decision? What can we learn about how our procedures and training actually perform under pressure?
2. Understand Work-as-Done, Not Just Work-as-Imagined
There’s always a gap between procedures (work-as-imagined) and actual practice (work-as-done). Understanding this gap and why it exists is more valuable than trying to force compliance with unrealistic procedures. Some of the most important quality improvements I’ve implemented came from understanding how operators actually solve problems under real conditions.
3. Measure Positive Capacities, Not Just Negative Events
Traditional quality metrics focus on what didn’t happen—no deviations, no complaints, no failures. I’ve started developing metrics around investigation quality, learning effectiveness, and adaptive capacity rather than just counting problems. How quickly do we identify and respond to emerging issues? How effectively do we share learning across sites? How well do our people handle unexpected situations?
4. Create Psychological Safety for Learning
Fear and punishment shut down the flow of safety-critical information. Organizations that want to learn from failures must create conditions where people can report problems, admit mistakes, and share concerns without fear of retribution. This is particularly challenging in our regulated environment, but it’s essential for moving beyond compliance theater toward genuine learning.
5. Focus on Contributing Conditions, Not Root Causes
Complex failures emerge from multiple interacting factors, not single root causes. The take-the-best approach I’ve been developing helps identify the most causally powerful factor while avoiding the trap of seeking THE cause. Understanding mechanisms is more valuable than finding someone to blame.
6. Embrace Adaptive Capacity Instead of Fighting Variability
People’s ability to adapt and respond to unexpected conditions is what makes complex systems work, not a threat to be controlled. Rather than trying to eliminate human variability through ever-more-prescriptive procedures, we should understand how that variability creates resilience and design systems that support rather than constrain adaptive problem-solving.
Connection to Investigation Excellence
Dekker’s work provides the theoretical foundation for many approaches I’ve been exploring on this blog. His emphasis on testable hypotheses rather than compliance theater directly supports falsifiable quality systems. His new view framework underlies the causal reasoning methods I’ve been developing. His focus on understanding normal work, not just failures, informs my approach to risk management.
Most importantly, his insistence on moving beyond negative reasoning (“what didn’t happen”) to positive causal statements (“what actually happened and why”) has transformed how I approach investigations. Instead of documenting failures to follow procedures, we’re understanding the specific mechanisms that drove events—and that makes all the difference in preventing recurrence.
Essential Reading for Quality Leaders
If you’re leading quality organizations in today’s complex regulatory environment, these Dekker works are essential:
Dekker’s work challenges us as quality leaders to move beyond the comfortable certainty of compliance-focused approaches toward the more demanding work of creating genuine learning systems. This requires admitting that our procedures and training might not work as intended. It means supporting people when they make mistakes rather than just punishing them. It demands that we measure our success by how well we learn and adapt, not just how well we document compliance.
This isn’t easy work. It requires the kind of organizational humility that Amy Edmondson and other leadership researchers emphasize—the willingness to be proven wrong in service of getting better. But in my experience, organizations that embrace this challenge develop more robust quality systems and, ultimately, better outcomes for patients.
The question isn’t whether Sidney Dekker is right about everything—it’s whether we’re willing to test his ideas and learn from the results. That’s exactly the kind of falsifiable approach that both his work and effective quality systems demand.
The draft Annex 11 is a cultural shift, a new way of working that reaches beyond pure compliance to emphasize accountability, transparency, and full-system oversight. Section 5.1, simply titled: “Cooperation” is a small but might part of this transformation
On its face, Section 5.1 may sound like a pleasantry: the regulation states that “there should be close cooperation between all relevant personnel such as process owner, system owner, qualified persons and IT.” In reality, this is a direct call to action for the formation of empowered, cross-functional, and highly integrated governance structures. It’s a recognition that, in an era when computerized systems underpin everything from batch release to deviation investigation, a siloed or transactional approach to system ownership is organizational malpractice.
Governance: From Siloed Ownership to Shared Accountability
Let’s breakdown what “cooperation” truly means in the current pharmaceutical digital landscape. Governance in the Annex 11 context is no longer a paperwork obligation but the backbone for digital trust. The roles of Process Owner (who understands the GMP-critical process), System Owner (managing the integrity and availability of the system), Quality (bearing regulatory release or oversight risk), and the IT function (delivering the technical and cybersecurity expertise) all must be clearly defined, actively engaged, and jointly responsible for compliance outcomes.
This shared ownership translates directly into how organizations structure project teams. Legacy models—where IT “owns the system,” Quality “owns compliance,” and business users “just use the tool”—are explicitly outdated. Section 5.1 obligates that these domains work in seamless partnership, not simply at “handover” moments but throughout every lifecycle phase from selection and implementation to maintenance and retirement. Each group brings indispensable knowledge: the process owner knows process risks and requirements; the system owner manages configuration and operational sustainability; Quality interprets regulatory standards and ensure release integrity; IT enables security, continuity, and technical change.
Practical Project Realities: Embedding Cooperation in Every Phase
In my experience, the biggest compliance failures often do not hinge on technical platform choices, but on fractured or missing cross-functional cooperation. Robust governance, under Section 5.1, doesn’t just mean having an org chart—it means everyone understands and fulfills their operational and compliance obligations every day. In practice, this requires formal documents (RACI matrices, governance charters), clear escalation routes, and regular—preferably, structured—forums for project and system performance review.
During system implementation, deep cooperation means all stakeholders are involved in requirements gathering and risk assessment, not just as “signatories” but as active contributors. It is not enough for the business to hand off requirements to IT with minimal dialogue, nor for IT to configure a system and expect the Qulity sign-off at the end. Instead, expect joint workshops, shared risk assessments (tying from process hazard analysis to technical configuration), and iterative reviews where each stakeholder is empowered to raise objections or demand proof of controls.
At all times, communication must be systematic, not ad hoc: regular governance meetings, with pre-published minutes and action tracking; dashboards or portals where issues, risks, and enhancement requests can be logged, tracked, and addressed; and shared access to documentation, validation reports, CAPA records, and system audit trails. This is particularly crucial as digital systems (cloud-based, SaaS, hybrid) increasingly blur the lines between “IT” and “business” roles.
Training, Qualifications, and Role Clarity: Everyone Is Accountable
Section 5.1 further clarifies that relevant personnel—regardless of functional home—must possess the appropriate qualifications, documented access rights, and clearly defined responsibilities. This raises the bar on both onboarding and continuing education. “Cooperation” thus demands rotational training and knowledge-sharing among core team members. Process owners must understand enough of IT and validation to foresee configuration-related compliance risks. IT staff must be fluent in GMP requirements and data integrity. Quality must move beyond audit response and actively participate in system configuration choices, validation planning, and periodic review.
In my own project experience, the difference between a successful, inspection-ready implementation and a troubled, remediation-prone rollout is almost always the presence, or absence, of this cross-trained, truly cooperative project team.
Supplier and Service Provider Partnerships: Extending Governance Beyond the Walls
The rise of cloud, SaaS, and outsourced system management means that “cooperation” extends outside traditional organizational boundaries. Section 5.1 works in concert with supplier sections of Annex 11—everyone from IT support to critical SaaS vendors must be engaged as partners within the governance framework. This requires clear, enforceable contracts outlining roles and responsibilities for security, data integrity, backup, and business continuity. It also means periodic supplier reviews, joint planning sessions, and supplier participation in incidents and change management when systems span organizations.
Internal IT must also be treated with the same rigor—a department supporting a GMP system is, under regulation, no different than a third-party vendor; it must be a named party in the cooperation and governance ecosystem.
Oversight and Monitoring: Governance as a Living Process
Effective cooperation isn’t a “set and forget”—it requires active, joint oversight. That means frequent management reviews (not just at system launch but periodically throughout the lifecycle), candid CAPA root cause debriefs across teams, and ongoing risk and performance evaluations done collectively. Each member of the governance body—be they system owner, process owner, or Quality—should have the right to escalate issues and trigger review of system configuration, validation status, or supplier contracts.
Structured communication frameworks—regularly scheduled project or operations reviews, joint documentation updates, and cross-functional risk and performance dashboards—turn this principle into practice. This is how validation, data integrity, and operational performance are confidently sustained (not just checked once) in a rigorous, documented, and inspection-ready fashion.
The “Cooperation” Imperative and the Digital GMP Transformation
With the explosion of digital complexity—artificial intelligence, platform integrations, distributed teams—the management of computerized systems has evolved well beyond technical mastery or GMP box-ticking. True compliance, under the new Annex 11, hangs on the ability of organizations to operationalize interdisciplinary governance. Section 5.1 thus becomes a proxy for digital maturity: teams that still operate in silos or treat “cooperation” as a formality will be missed by the first regulatory deep dive or major incident.
Meanwhile, sites that embed clear role assignment, foster cross-disciplinary partnership, and create active, transparent governance processes (documented and tracked) will find not only that inspections run smoothly—they’ll spend less time in audit firefighting, make faster decisions during technology rollouts, and spot improvement opportunities early.
Teams that embrace the cooperation mandate see risk mitigation, continuous improvement, and regulatory trust as the natural byproducts of shared accountability. Those that don’t will find themselves either in chronic remediation or watching more agile, digitally mature competitors pull ahead.
Key Governance and Project Team Implications
To provide a summary for project, governance, and operational leaders, here is a table distilling the new paradigm:
Governance Aspect
Implications for Project & Governance Teams
Clear Role Assignment
Define and document responsibilities for process owners, system owners, and IT.
Cross-Functional Partnership
Ensure collaboration among quality, IT, validation, and operational teams.
Training & Qualification
Clarify required qualifications, access levels, and competencies for personnel.
Supplier Oversight
Establish contracts with roles, responsibilities, and audit access rights.
Proactive Monitoring
Maintain joint oversight mechanisms to promptly address issues and changes.
Communication Framework
Set up regular, documented interaction channels among involved stakeholders.
In this new landscape, “cooperation” is not a regulatory afterthought. It is the hinge on which the entire digital validation and integrity culture swings. How and how well your teams work together is now as much a matter of inspection and business success as any technical control, risk assessment, or test script.
The draft EU Annex 11 Section 11 “Identity and Access Management” reads like a complete demolition of every lazy access-control practice organizations might have been coasting on for years. Gone are the vague handwaves about “appropriate controls.” The new IAM requirements are explicitly designed to eliminate the shared-account shortcuts and password recycling schemes that have made pharma IT security a running joke among auditors.
The regulatory bar for access management has been raised so high that most existing computerized systems will need major overhauls to comply. Organizations that think a username-password combo and quarterly access reviews will satisfy the new requirements are about to learn some expensive lessons about modern data integrity enforcement.
What Makes This Different from Every Other Access Control Update
The draft Annex 11’s Identity and Access Management section is a complete philosophical shift from “trust but verify” to “prove everything, always.” Where the 2011 version offered generic statements about restricting access to “authorised persons,” the 2025 draft delivers 11 detailed subsections that read like a cybersecurity playbook written by paranoid auditors who’ve spent too much time investigating data integrity failures.
This isn’t incremental improvement. Section 11 transforms IAM from a compliance checkbox into a fundamental pillar of data integrity that touches every aspect of how users interact with GMP systems. The draft makes it explicitly clear that poor access controls are considered violations of data integrity—not just security oversights.
European regulators have decided that the EU needs robust—and arguably more prescriptive—guidance for managing user access in an era where cloud services, remote work, and cyber threats have fundamentally changed the risk landscape. The result is regulatory text that assumes bad actors, compromised credentials, and insider threats as baseline conditions rather than edge cases.
The Eleven Subsections That Will Break Your Current Processes
11.1: Unique Accounts – The Death of Shared Logins
The draft opens with a declaration that will send shivers through organizations still using shared service accounts: “All users should have unique and personal accounts. The use of shared accounts except for those limited to read-only access (no data or settings can be changed), constitute a violation of data integrity”.
This isn’t a suggestion—it’s a flat prohibition with explicit regulatory consequences. Every shared “QC_User” account, every “Production_Shift” login, every “Maintenance_Team” credential becomes a data integrity violation the moment this guidance takes effect. The only exception is read-only accounts that cannot modify data or settings, which means most shared accounts used for batch record reviews, approval workflows, and system maintenance will need complete redesign.
The impact extends beyond just creating more user accounts. This sets out the need to address all the legacy systems that have coasted along for years. There are a lot of filter integrity testers, pH meters and balances, among other systems, that will require some deep views.
Where the 2011 Annex 11 simply required that access changes “should be recorded,” the draft demands “continuous management” with timely granting, modification, and revocation as users “join, change, and end their involvement in GMP activities”. The word “timely” appears to be doing significant regulatory work here—expect inspectors to scrutinize how quickly access is updated when employees change roles or leave the organization.
This requirement acknowledges the reality that modern pharmaceutical operations involve constant personnel changes, contractor rotations, and cross-functional project teams. Static annual access reviews become insufficient when users need different permissions for different projects, temporary elevated access for system maintenance, and immediate revocation when employment status changes. The continuous management standard implies real-time or near-real-time access administration that most organizations currently lack.
The operational implications are clear. It is no longer optional not to integrate HR systems with IT provisioning tools and tie it into your GxP systems. Contractor management processes will require pre-defined access templates and automatic expiration dates. Organizations that treat access management as a periodic administrative task rather than a dynamic business process will find themselves fundamentally out of compliance.
11.3: Certain Identification – The End of Token-Only Authentication
Perhaps the most technically disruptive requirement, Section 11.3 mandates authentication methods that “identify users with a high degree of certainty” while explicitly prohibiting “authentication only by means of a token or a smart card…if this could be used by another user”. This effectively eliminates proximity cards, USB tokens, and other “something you have” authentication methods as standalone solutions.
The regulation acknowledges biometric authentication as acceptable but requires username and password as the baseline, with other methods providing “at least the same level of security”. For organizations that have invested heavily in smart card infrastructure or hardware tokens, this represents a significant technology shift toward multi-factor authentication combining knowledge and possession factors.
The “high degree of certainty” language introduces a subjective standard that will likely be interpreted differently across regulatory jurisdictions. Organizations should expect inspectors to challenge any authentication method that could reasonably be shared, borrowed, or transferred between users. This standard effectively rules out any authentication approach that doesn’t require active user participation—no more swiping someone else’s badge to help them log in during busy periods.
Biometric systems become attractive under this standard, but the draft doesn’t provide guidance on acceptable biometric modalities, error rates, or privacy considerations. Organizations implementing fingerprint, facial recognition, or voice authentication systems will need to document the security characteristics that meet the “high degree of certainty” requirement while navigating European privacy regulations that may restrict biometric data collection.
11.4: Confidential Passwords – Personal Responsibility Meets System Enforcement
The draft’s password confidentiality requirements combine personal responsibility with system enforcement in ways that current pharmaceutical IT environments rarely support. Section 11.4 requires passwords to be “kept confidential and protected from all other users, both at system and at a personal level” while mandating that “passwords received from e.g. a manager, or a system administrator should be changed at the first login, preferably required by the system”1.
This requirement targets the common practice of IT administrators assigning temporary passwords that users may or may not change, creating audit trail ambiguity about who actually performed specific actions. The “preferably required by the system” language suggests that technical controls should enforce password changes rather than relying on user compliance with written procedures.
The personal responsibility aspect extends beyond individual users to organizational accountability. Companies must demonstrate that their password policies, training programs, and technical controls work together to prevent password sharing, writing passwords down, or other practices that compromise authentication integrity. This creates a documentation burden for organizations to prove that their password management practices support data integrity objectives.
11.5: Secure Passwords – Risk-Based Complexity That Actually Works
Rather than mandating specific password requirements, Section 11.5 takes a risk-based approach that requires password rules to be “commensurate with risks and consequences of unauthorised changes in systems and data”. For critical systems, the draft specifies passwords should be “of sufficient length to effectively prevent unauthorised access and contain a combination of uppercase, lowercase, numbers and symbols”.
The regulation prohibits common password anti-patterns: “A password should not contain e.g. words that can be found in a dictionary, the name of a person, a user id, product or organisation, and should be significantly different from a previous password”. This requirement goes beyond basic complexity rules to address predictable password patterns that reduce security effectiveness.
The risk-based approach means organizations must document their password requirements based on system criticality assessments. Manufacturing control systems, quality management databases, and regulatory submission platforms may require different password standards than training systems or general productivity applications. This creates a classification burden where organizations must justify their password requirements through formal risk assessments.
“Sufficient length” and “significantly different” introduce subjective standards that organizations must define and defend. Expect regulatory discussions about whether 8-character passwords meet the “sufficient length” requirement for critical systems, and whether changing a single character constitutes “significantly different” from previous passwords.
11.6: Strong Authentication – MFA for Remote Access
Section 11.6 represents the draft’s most explicit cybersecurity requirement: “Remote authentication on critical systems from outside controlled perimeters, should include multifactor authentication (MFA)”. This requirement acknowledges the reality of remote work, cloud services, and mobile access to pharmaceutical systems while establishing clear security expectations.
The “controlled perimeters” language requires organizations to define their network security boundaries and distinguish between internal and external access. Users connecting from corporate offices, manufacturing facilities, and other company-controlled locations may use different authentication methods than those connecting from home, hotels, or public networks.
“Critical systems” must be defined through risk assessment processes, creating another classification requirement. Organizations must identify which systems require MFA for remote access and document the criteria used for this determination. Laboratory instruments, manufacturing equipment, and quality management systems will likely qualify as critical, but organizations must make these determinations explicitly.
The MFA requirement doesn’t specify acceptable second factors, leaving organizations to choose between SMS codes, authenticator applications, hardware tokens, biometric verification, or other technologies. However, the emphasis on security effectiveness suggests that easily compromised methods like SMS may not satisfy regulatory expectations for critical system access.
11.7: Auto Locking – Administrative Controls for Security Failures
Account lockout requirements in Section 11.7 combine automated security controls with administrative oversight in ways that current pharmaceutical systems rarely implement effectively. The draft requires accounts to be “automatically locked after a pre-defined number of successive failed authentication attempts” with “accounts should only be unlocked by the system administrator after it has been confirmed that this was not part of an unauthorised login attempt or after the risk for such attempt has been removed”.
This requirement transforms routine password lockouts from simple user inconvenience into formal security incident investigations. System administrators cannot simply unlock accounts upon user request—they must investigate the failed login attempts and document their findings before restoring access. For organizations with hundreds or thousands of users, this represents a significant administrative burden that requires defined procedures and potentially additional staffing.
The “pre-defined number” must be established through risk assessment and documented in system configuration. Three failed attempts may be appropriate for highly sensitive systems, while five or more attempts might be acceptable for lower-risk applications. Organizations must justify their lockout thresholds based on balancing security protection with operational efficiency.
“Unauthorised login attempt” investigations require forensic capabilities that many pharmaceutical IT organizations currently lack. System administrators must be able to analyze login patterns, identify potential attack signatures, and distinguish between user errors and malicious activity. This capability implies enhanced logging, monitoring tools, and security expertise that extends beyond traditional IT support functions.
11.8: Inactivity Logout – Session Management That Users Cannot Override
Session management requirements in Section 11.8 establish mandatory timeout controls that users cannot circumvent: “Systems should include an automatic inactivity logout, which logs out a user after a defined period of inactivity. The user should not be able to change the inactivity logout time (outside defined and acceptable limits) or deactivate the functionality”.
The requirement for re-authentication after inactivity logout means users cannot simply resume their sessions—they must provide credentials again, creating multiple authentication points throughout extended work sessions. This approach prevents unauthorized access to unattended workstations while ensuring that long-running analytical procedures or batch processing operations don’t compromise security.
“Defined and acceptable limits” requires organizations to establish timeout parameters based on risk assessment while potentially allowing users some flexibility within security boundaries. A five-minute timeout might be appropriate for systems that directly impact product quality, while 30-minute timeouts could be acceptable for documentation or training applications.
The prohibition on user modification of timeout settings eliminates common workarounds where users extend session timeouts to avoid frequent re-authentication. System configurations must enforce these settings at a level that prevents user modification, requiring administrative control over session management parameters.
Section 11.9 establishes detailed logging requirements that extend far beyond basic audit trail functionality: “Systems should include an access log (separate, or as part of the audit trail) which, for each login, automatically logs the username, user role (if possible, to choose between several roles), the date and time for login, the date and time for logout (incl. inactivity logout)”.
The “separate, or as part of the audit trail” language recognizes that authentication events may need distinct handling from data modification events. Organizations must decide whether to integrate access logs with existing audit trail systems or maintain separate authentication logging capabilities. This decision affects log analysis, retention policies, and regulatory presentation during inspections.
Role logging requirements are particularly significant for organizations using role-based access control systems. Users who can assume different roles during a session (QC analyst, batch reviewer, system administrator) must have their role selections logged with each login event. This requirement supports accountability by ensuring auditors can determine which permissions were active during specific time periods.
The requirement for logs to be “sortable and searchable, or alternatively…exported to a tool which provides this functionality” establishes performance standards for authentication logging systems. Organizations cannot simply capture access events—they must provide analytical capabilities that support investigation, trend analysis, and regulatory review.
11.10: Guiding Principles – Segregation of Duties and Least Privilege
Section 11.10 codifies two fundamental security principles that transform access management from user convenience to risk mitigation: “Segregation of duties, i.e. that users who are involved in GMP activities do not have administrative privileges” and “Least privilege principle, i.e. that users do not have higher access privileges than what is necessary for their job function”.
Segregation of duties eliminates the common practice of granting administrative rights to power users, subject matter experts, or senior personnel who also perform GMP activities. Quality managers cannot also serve as system administrators. Production supervisors cannot have database administrative privileges. Laboratory directors cannot configure their own LIMS access permissions. This separation requires organizations to maintain distinct IT support functions independent from GMP operations.
The least privilege principle requires ongoing access optimization rather than one-time role assignments. Users should receive minimum necessary permissions for their specific job functions, with temporary elevation only when required for specific tasks. This approach conflicts with traditional pharmaceutical access management where users often accumulate permissions over time or receive broad access to minimize support requests.
Implementation of these principles requires formal role definition, access classification, and privilege escalation procedures. Organizations must document job functions, identify minimum necessary permissions, and establish processes for temporary access elevation when users need additional capabilities for specific projects or maintenance activities.
The final requirement establishes ongoing access governance through “recurrent reviews where managers confirm the continued access of their employees in order to detect accesses which should have been changed or revoked during daily operation, but were accidentally forgotten”. This requirement goes beyond periodic access reviews to establish manager accountability for their team’s system permissions.
Manager confirmation creates personal responsibility for access accuracy rather than delegating reviews to IT or security teams. Functional managers must understand what systems their employees access, why those permissions are necessary, and whether access levels remain appropriate for current job responsibilities. This approach requires manager training on system capabilities and access implications.
Role-based access reviews extend the requirement to organizational roles rather than just individual users: “If user accounts are managed by means of roles, these should be subject to the same kind of reviews, where the accesses of roles are confirmed”. Organizations using role-based access control must review role definitions, permission assignments, and user-to-role mappings with the same rigor applied to individual account reviews.
Documentation and action requirements ensure that reviews produce evidence and corrections: “The reviews should be documented, and appropriate action taken”. Organizations cannot simply perform reviews—they must record findings, document decisions, and implement access modifications identified during the review process.
Risk-based frequency allows organizations to adjust review cycles based on system criticality: “The frequency of these reviews should be commensurate with the risks and consequences of changes in systems and data made by unauthorised individuals”. Critical manufacturing systems may require monthly reviews, while training systems might be reviewed annually.
How This Compares to 21 CFR Part 11 and Current Best Practices
The draft Annex 11’s Identity and Access Management requirements represent a significant advancement over 21 CFR Part 11, which addressed access control through basic authority checks and user authentication rather than comprehensive identity management. Part 11’s requirement for “at least two distinct identification components” becomes the foundation for much more sophisticated authentication and access control frameworks.
Multi-factor authentication requirements in the draft Annex 11 exceed Part 11 expectations by mandating MFA for remote access to critical systems, while Part 11 remains silent on multi-factor approaches. This difference reflects 25 years of cybersecurity evolution and acknowledges that username-password combinations provide insufficient protection for modern threat environments.
Current data integrity best practices have evolved toward comprehensive access management, risk-based authentication, and continuous monitoring—approaches that the draft Annex 11 now mandates rather than recommends. Organizations following ALCOA+ principles and implementing robust access controls will find the new requirements align with existing practices, while those relying on minimal compliance approaches will face significant gaps.
The Operational Reality of Implementation
System Architecture Changes
Most pharmaceutical computerized systems were designed assuming manual access management and periodic reviews would satisfy regulatory requirements. The draft Annex 11 requirements will force fundamental architecture changes including:
Identity Management Integration: Manufacturing execution systems, laboratory information management systems, and quality management platforms must integrate with centralized identity management systems to support continuous access management and role-based controls.
Authentication Infrastructure: Organizations must deploy multi-factor authentication systems capable of supporting diverse user populations, remote access scenarios, and integration with existing applications.
Logging and Monitoring: Enhanced access logging requirements demand centralized log management, analytical capabilities, and integration between authentication systems and audit trail infrastructure.
Session Management: Applications must implement configurable session timeout controls, prevent user modification of security settings, and support re-authentication without disrupting long-running processes.
Process Reengineering Requirements
The regulatory requirements will force organizations to redesign fundamental access management processes:
Continuous Provisioning: HR onboarding, role changes, and termination processes must trigger immediate access modifications rather than waiting for periodic reviews.
Manager Accountability: Access review processes must shift from IT-driven activities to manager-driven confirmations with documented decision-making and corrective actions.
Risk-Based Classification: Organizations must classify systems based on criticality, define access requirements accordingly, and maintain documentation supporting these determinations.
Incident Response: Account lockout events must trigger formal security investigations rather than simple password resets, requiring enhanced forensic capabilities and documented procedures.
Manager Training: Functional managers must understand system capabilities, access implications, and review responsibilities rather than delegating access decisions to IT teams.
User Education: Password security, MFA usage, and session management practices require user training programs that emphasize data integrity implications rather than just security compliance.
IT Skill Development: System administrators must develop security investigation capabilities, risk assessment skills, and regulatory compliance expertise beyond traditional technical support functions.
Audit Readiness: Organizations must prepare to demonstrate access control effectiveness through documentation, metrics, and investigative capabilities during regulatory inspections.
Strategic Implementation Approach
The Annex 11 Draft is just taking good cybersecurity and enshrining it more firmly in the GMPs. Organizations should not wait for the effective version to implement. Get that budget prioritized and start now.
Phase 1: Assessment and Classification
Organizations should begin with comprehensive assessment of current access control practices against the new requirements:
System Inventory: Catalog all computerized systems used in GMP activities, identifying shared accounts, authentication methods, and access control capabilities.
Risk Classification: Determine which systems qualify as “critical” requiring enhanced authentication and access controls.
Gap Analysis: Compare current practices against each subsection requirement, identifying technical, procedural, and training gaps.
Compliance Timeline: Develop implementation roadmap aligned with expected regulatory effective dates and system upgrade cycles.
Phase 2: Infrastructure Development
Focus on foundational technology changes required to support the new requirements:
Identity Management Platform: Deploy or enhance centralized identity management systems capable of supporting continuous provisioning and role-based access control.
Multi-Factor Authentication: Implement MFA systems supporting diverse authentication methods and integration with existing applications.
Enhanced Logging: Deploy log management platforms capable of aggregating, analyzing, and presenting access events from distributed systems.
Session Management: Upgrade applications to support configurable timeout controls and prevent user modification of security settings.
Phase 3: Process Implementation
Redesign access management processes to support continuous management and enhanced accountability:
Provisioning Automation: Integrate HR systems with IT provisioning tools to support automatic access changes based on employment events.
Manager Accountability: Train functional managers on access review responsibilities and implement documented review processes.
Security Incident Response: Develop procedures for investigating account lockouts and documenting security findings.
Audit Trail Integration: Ensure access events are properly integrated with existing audit trail review and batch release processes.
Phase 4: Validation and Documentation
When the Draft becomes effective you’ll be ready to complete validation activities demonstrating compliance with the new requirements:
Access Control Testing: Validate that technical controls prevent unauthorized access, enforce authentication requirements, and log security events appropriately.
Process Verification: Demonstrate that access management processes support continuous management, manager accountability, and risk-based reviews.
Training Verification: Document that personnel understand their responsibilities for password security, session management, and access control compliance.
Audit Readiness: Prepare documentation, metrics, and investigative capabilities required to demonstrate compliance during regulatory inspections.
The Competitive Advantage of Early Implementation
Organizations that proactively implement the draft Annex 11 IAM requirements will gain significant advantages beyond regulatory compliance:
Enhanced Security Posture: The access control improvements provide protection against cyber threats, insider risks, and accidental data compromise that extend beyond GMP applications to general IT security.
Operational Efficiency: Automated provisioning, role-based access, and centralized identity management reduce administrative overhead while improving access accuracy.
Audit Confidence: Comprehensive access logging, manager accountability, and continuous management provide evidence of control effectiveness that regulators and auditors will recognize.
Digital Transformation Enablement: Modern identity and access management infrastructure supports cloud adoption, mobile access, and advanced analytics initiatives that drive business value.
Risk Mitigation: Enhanced access controls reduce the likelihood of data integrity violations, security incidents, and regulatory findings that can disrupt operations and damage reputation.
Looking Forward: The End of Security Theater
The draft Annex 11’s Identity and Access Management requirements represent the end of security theater in pharmaceutical access control. Organizations can no longer satisfy regulatory expectations through generic policies and a reliance on periodic reviews to provide the appearance of control without actual security effectiveness.
The new requirements assume that user access is a continuous risk requiring active management, real-time monitoring, and ongoing verification. This approach aligns with modern cybersecurity practices while establishing regulatory expectations that reflect the actual threat environment facing pharmaceutical operations.
Implementation success will require significant investment in technology infrastructure, process reengineering, and organizational change management. However, organizations that embrace these requirements as opportunities for security improvement rather than compliance burdens will build competitive advantages that extend far beyond regulatory satisfaction.
The transition period between now and the expected 2026 effective date provides a ideal window for organizations to assess their current practices, develop implementation strategies, and begin the technical and procedural changes required for compliance. Organizations that delay implementation risk finding themselves scrambling to achieve compliance while their competitors demonstrate regulatory leadership through proactive adoption.
For pharmaceutical organizations serious about data integrity, operational security, and regulatory compliance, the draft Annex 11 IAM requirements aren’t obstacles to overcome—they’re the roadmap to building access control practices worthy of the products and patients we serve. The only question is whether your organization will lead this transformation or follow in the wake of those who do.
Requirement
Current Annex 11 (2011)
Draft Annex 11 Section 11 (2025)
21 CFR Part 11
User Account Management
Basic – creation, change, cancellation should be recorded
Continuous management – grant, modify, revoke as users join/change/leave
Basic user management, creation/change/cancellation recorded
Authentication Methods
Physical/logical controls, pass cards, personal codes with passwords, biometrics
Username + password or equivalent (biometrics); tokens/smart cards alone insufficient
At least two distinct identification components (ID code + password)
Password Requirements
Not specified in detail
Secure passwords enforced by systems, length/complexity based on risk, dictionary words prohibited
The Hidden Architecture of Risk Assessment Failure
Peter Baker‘s blunt assessment, “We allowed all these players into the market who never should have been there in the first place, ” hits at something we all recognize but rarely talk about openly. Here’s the uncomfortable truth: even seasoned quality professionals with decades of experience and proven methodologies can miss critical risks that seem obvious in hindsight. Recognizing this truth is not about competence or dedication. It is about acknowledging that our expertise, no matter how extensive, operates within cognitive frameworks that can create blind spots. The real opportunity lies in understanding how these mental patterns shape our decisions and building knowledge systems that help us see what we might otherwise miss. When we’re honest about these limitations, we can strengthen our approaches and create more robust quality systems.
The framework of risk management, designed to help avoid the monsters of bad decision-making, can all too often fail us. Luckily, the Pharmaceutical Inspection Co-operation Scheme (PIC/S) guidance document PI 038-2 “Assessment of Quality Risk Management Implementation” identifies three critical observations that reveal systematic vulnerabilities in risk management practice: unjustified assumptions, incomplete identification of risks or inadequate information, and lack of relevant experience with inappropriate use of risk assessment tools. These observations represent something more profound than procedural failures—they expose cognitive and knowledge management vulnerabilities that can undermine even the most well-intentioned quality systems..
Understanding these vulnerabilities through the lens of cognitive behavioral science and knowledge management principles provides a pathway to more robust and resilient quality systems. Instead of viewing these failures as isolated incidents or individual shortcomings, we should recognize them as predictable patterns that emerge from systematic limitations in how humans process information and organizations manage knowledge. This recognition opens the door to designing quality systems that work with, rather than against, these cognitive realities
The Framework Foundation of Risk Management Excellence
Risk management operates fundamentally as a frameworkrather than a rigid methodology, providing the structural architecture that enables systematic approaches to identifying, assessing, and controlling uncertainties that could impact pharmaceutical quality objectives. This distinction proves crucial for understanding how cognitive biases manifest within risk management systems and how excellence-driven quality systems can effectively address them.
A framework establishes the high-level structure, principles, and processes for managing risks systematically while allowing flexibility in execution and adaptation to specific organizational contexts. The framework defines structural components like governance and culture, strategy and objective-setting, and performance monitoring that establish the scaffolding for risk management without prescribing inflexible procedures.
Within this framework structure, organizations deploy specific methodological elements as tools for executing particular risk management tasks. These methodologies include techniques such as Failure Mode and Effects Analysis (FMEA), brainstorming sessions, SWOT analysis, and risk surveys for identification activities, while assessment methodologies encompass qualitative and quantitative approaches including statistical models and scenario analysis. The critical insight is that frameworks provide the systematic architecture that counters cognitive biases, while methodologies are specific techniques deployed within this structure.
This framework approach directly addresses the three PIC/S observations by establishing systematic requirements that counter natural cognitive tendencies. Standardized framework processes force systematic consideration of risk factors rather than allowing teams to rely on intuitive pattern recognition that might be influenced by availability bias or anchoring on familiar scenarios. Documented decision rationales required by framework approaches make assumptions explicit and subject to challenge, preventing the perpetuation of unjustified beliefs that may have become embedded in organizational practices.
The governance components inherent in risk management frameworks address the expertise and knowledge management challenges identified in PIC/S guidance by establishing clear roles, responsibilities, and requirements for appropriate expertise involvement in risk assessment activities. Rather than leaving expertise requirements to chance or individual judgment, frameworks systematically define when specialized knowledge is required and how it should be accessed and validated.
ICH Q9’s approach to Quality Risk Management in pharmaceuticals demonstrates this framework principle through its emphasis on scientific knowledge and proportionate formality. The guideline establishes framework requirements that risk assessments be “based on scientific knowledge and linked to patient protection” while allowing methodological flexibility in how these requirements are met. This framework approach provides systematic protection against the cognitive biases that lead to unjustified assumptions while supporting the knowledge management processes necessary for complete risk identification and appropriate tool application.
The continuous improvement cycles embedded in mature risk management frameworks provide ongoing validation of cognitive bias mitigation effectiveness through operational performance data. These systematic feedback loops enable organizations to identify when initial assumptions prove incorrect or when changing conditions alter risk profiles, supporting the adaptive learning required for sustained excellence in pharmaceutical risk management.
The Systematic Nature of Risk Assessment Failure
Unjustified Assumptions: When Experience Becomes Liability
The first PIC/S observation—unjustified assumptions—represents perhaps the most insidious failure mode in pharmaceutical risk management. These are decisions made without sufficient scientific evidence or rational basis, often arising from what appears to be strength: extensive experience with familiar processes. The irony is that the very expertise we rely upon can become a source of systematic error when it leads to unfounded confidence in our understanding.
This phenomenon manifests most clearly in what cognitive scientists call anchoring bias—the tendency to rely too heavily on the first piece of information encountered when making decisions. In pharmaceutical risk assessments, this might appear as teams anchoring on historical performance data without adequately considering how process changes, equipment aging, or supply chain modifications might alter risk profiles. The assumption becomes: “This process has worked safely for five years, so the risk profile remains unchanged.”
Confirmation bias compounds this issue by causing assessors to seek information that confirms their existing beliefs while ignoring contradictory evidence. Teams may unconsciously filter available data to support predetermined conclusions about process reliability or control effectiveness. This creates a self-reinforcing cycle where assumptions become accepted facts, protected from challenge by selective attention to supporting evidence.
The knowledge management dimension of this failure is equally significant. Organizations often lack systematic approaches to capturing and validating the assumptions embedded in institutional knowledge. Tacit knowledge—the experiential, intuitive understanding that experts develop over time—becomes problematic when it remains unexamined and unchallenged. Without explicit processes to surface and test these assumptions, they become invisible constraints on risk assessment effectiveness.
Incomplete Risk Identification: The Boundaries of Awareness
The second observation—incomplete identification of risks or inadequate information—reflects systematic failures in the scope and depth of risk assessment activities. This represents more than simple oversight; it demonstrates how cognitive limitations and organizational boundaries constrain our ability to identify potential hazards comprehensively.
Availability bias plays a central role in this failure mode. Risk assessment teams naturally focus on hazards that are easily recalled or recently experienced, leading to overemphasis on dramatic but unlikely events while underestimating more probable but less memorable risks. A team might spend considerable time analyzing the risk of catastrophic equipment failure while overlooking the cumulative impact of gradual process drift or material variability.
The knowledge management implications are profound. Organizations often struggle with knowledge that exists in isolated pockets of expertise. Critical information about process behaviors, failure modes, or control limitations may be trapped within specific functional areas or individual experts. Without systematic mechanisms to aggregate and synthesize distributed knowledge, risk assessments operate on fundamentally incomplete information.
Groupthink and organizational boundaries further constrain risk identification. When risk assessment teams are composed of individuals from similar backgrounds or organizational levels, they may share common blind spots that prevent recognition of certain hazard categories. The pressure to reach consensus can suppress dissenting views that might identify overlooked risks.
Inappropriate Tool Application: When Methodology Becomes Mythology
The third observation—lack of relevant experience with process assessment and inappropriate use of risk assessment tools—reveals how methodological sophistication can mask fundamental misunderstanding. This failure mode is particularly dangerous because it generates false confidence in risk assessment conclusions while obscuring the limitations of the analysis.
Overconfidence bias drives teams to believe they have more expertise than they actually possess, leading to misapplication of complex risk assessment methodologies. A team might apply Failure Mode and Effects Analysis (FMEA) to a novel process without adequate understanding of either the methodology’s limitations or the process’s unique characteristics. The resulting analysis appears scientifically rigorous while providing misleading conclusions about risk levels and control effectiveness.
This connects directly to knowledge management failures in expertise distribution and access. Organizations may lack systematic approaches to identifying when specialized knowledge is required for risk assessments and ensuring that appropriate expertise is available when needed. The result is risk assessments conducted by well-intentioned teams who lack the specific knowledge required for accurate analysis.
The problem is compounded when organizations rely heavily on external consultants or standardized methodologies without developing internal capabilities for critical evaluation. While external expertise can be valuable, sole reliance on these resources may result in inappropriate conclusions or a lack of ownership of the assessment, as the PIC/S guidance explicitly warns.
The Role of Negative Reasoning in Risk Assessment
The research on causal reasoning versus negative reasoning from Energy Safety Canada provides additional insight into systematic failures in pharmaceutical risk assessments. Traditional root cause analysis often focuses on what did not happen rather than what actually occurred—identifying “counterfactuals” such as “operators not following procedures” or “personnel not stopping work when they should have.”
This approach, termed “negative reasoning,” is fundamentally flawed because what was not happening cannot create the outcomes we experienced. These counterfactuals “exist only in retrospection and never actually influenced events,” yet they dominate many investigation conclusions. In risk assessment contexts, this manifests as teams focusing on the absence of desired behaviors or controls rather than understanding the positive factors that actually influence system performance.
The shift toward causal reasoning requires understanding what actually occurred and what factors positively influenced the outcomes observed.
Knowledge-Enabled Decision Making
The intersection of cognitive science and knowledge management reveals how organizations can design systems that support better risk assessment decisions. Knowledge-enabled decision making requires structures that make relevant information accessible at the point of decision while supporting the cognitive processes necessary for accurate analysis.
This involves several key elements:
Structured knowledge capture that explicitly identifies assumptions, limitations, and context for recorded information. Rather than simply documenting conclusions, organizations must capture the reasoning process and evidence base that supports risk assessment decisions.
Knowledge validation systems that systematically test assumptions embedded in organizational knowledge. This includes processes for challenging accepted wisdom and updating mental models when new evidence emerges.
Expertise networks that connect decision-makers with relevant specialized knowledge when required. Rather than relying on generalist teams for all risk assessments, organizations need systematic approaches to accessing specialized expertise when process complexity or novelty demands it.
Decision support systems that prompt systematic consideration of potential biases and alternative explanations.
Excellence and Elegance: Designing Quality Systems for Cognitive Reality
Structured Decision-Making Processes
Excellence in pharmaceutical quality systems requires moving beyond hoping that individuals will overcome cognitive limitations through awareness alone. Instead, organizations must design structured decision-making processes that systematically counter known biases while supporting comprehensive risk identification and analysis.
Forced systematic consideration involves using checklists, templates, and protocols that require teams to address specific risk categories and evidence types before reaching conclusions. Rather than relying on free-form discussion that may be influenced by availability bias or groupthink, these tools ensure comprehensive coverage of relevant factors.
Devil’s advocate processes systematically introduce alternative perspectives and challenge preferred conclusions. By assigning specific individuals to argue against prevailing views or identify overlooked risks, organizations can counter confirmation bias and overconfidence while identifying blind spots in risk assessments.
Staged decision-making separates risk identification from risk evaluation, preventing premature closure and ensuring adequate time for comprehensive hazard identification before moving to analysis and control decisions.
Multi-Perspective Analysis and Diverse Assessment Teams
Cognitive diversity in risk assessment teams provides natural protection against individual and group biases. This goes beyond simple functional representation to include differences in experience, training, organizational level, and thinking styles that can identify risks and solutions that homogeneous teams might miss.
Cross-functional integration ensures that risk assessments benefit from different perspectives on process performance, control effectiveness, and potential failure modes. Manufacturing, quality assurance, regulatory affairs, and technical development professionals each bring different knowledge bases and mental models that can reveal different aspects of risk.
External perspectives through consultants, subject matter experts from other sites, or industry benchmarking can provide additional protection against organizational blind spots. However, as the PIC/S guidance emphasizes, these external resources should facilitate and advise rather than replace internal ownership and accountability.
Rotating team membership for ongoing risk assessment activities prevents the development of group biases and ensures fresh perspectives on familiar processes. This also supports knowledge transfer and prevents critical risk assessment capabilities from becoming concentrated in specific individuals.
Evidence-Based Analysis Requirements
Scientific justification for all risk assessment conclusions requires teams to base their analysis on objective, verifiable data rather than assumptions or intuitive judgments. This includes collecting comprehensive information about process performance, material characteristics, equipment reliability, and environmental factors before drawing conclusions about risk levels.
Assumption documentation makes implicit beliefs explicit and subject to challenge. Any assumptions made during risk assessment must be clearly identified, justified with available evidence, and flagged for future validation. This transparency helps identify areas where additional data collection may be needed and prevents assumptions from becoming accepted facts over time.
Evidence quality assessment evaluates the strength and reliability of information used to support risk assessment conclusions. This includes understanding limitations, uncertainties, and potential sources of bias in the data itself.
Structured uncertainty analysisexplicitly addresses areas where knowledge is incomplete or confidence is low. Rather than treating uncertainty as a weakness to be minimized, mature quality systems acknowledge uncertainty and design controls that remain effective despite incomplete information.
Continuous Monitoring and Reassessment Systems
Performance validation provides ongoing verification of risk assessment accuracy through operational performance data. The PIC/S guidance emphasizes that risk assessments should be “periodically reviewed for currency and effectiveness” with systems to track how well predicted risks align with actual experience.
Assumption testing uses operational data to validate or refute assumptions embedded in risk assessments. When monitoring reveals discrepancies between predicted and actual performance, this triggers systematic review of the original assessment to identify potential sources of bias or incomplete analysis.
Feedback loopsensure that lessons learned from risk assessment performance are incorporated into future assessments. This includes both successful risk predictions and instances where significant risks were initially overlooked.
Adaptive learning systems use accumulated experience to improve risk assessment methodologies and training programs. Organizations can track patterns in assessment effectiveness to identify systematic biases or knowledge gaps that require attention.
Knowledge Management as the Foundation of Cognitive Excellence
The Critical Challenge of Tacit Knowledge Capture
ICH Q10’s definition of knowledge management as “a systematic approach to acquiring, analysing, storing and disseminating information related to products, manufacturing processes and components” provides the regulatory framework, but the cognitive dimensions of knowledge management are equally critical. The distinction between tacit knowledge (experiential, intuitive understanding) and explicit knowledge (documented procedures and data) becomes crucial when designing systems to support effective risk assessment.
Tacit knowledge capture represents one of the most significant challenges in pharmaceutical quality systems. The experienced process engineer who can “feel” when a process is running correctly possesses invaluable knowledge, but this knowledge remains vulnerable to loss through retirements, organizational changes, or simply the passage of time. More critically, tacit knowledge often contains embedded assumptions that may become outdated as processes, materials, or environmental conditions change.
Structured knowledge elicitation processes systematically capture not just what experts know, but how they know it—the cues, patterns, and reasoning processes that guide their decision-making. This involves techniques such as cognitive interviewing, scenario-based discussions, and systematic documentation of decision rationales that make implicit knowledge explicit and subject to validation.
Knowledge validation and updating cycles ensure that captured knowledge remains current and accurate. This is particularly important for tacit knowledge, which may be based on historical conditions that no longer apply. Systematic processes for testing and updating knowledge prevent the accumulation of outdated assumptions that can compromise risk assessment effectiveness.
Expertise Distribution and Access
Knowledge networks provide systematic approaches to connecting decision-makers with relevant expertise when complex risk assessments require specialized knowledge. Rather than assuming that generalist teams can address all risk assessment challenges, mature organizations develop capabilities to identify when specialized expertise is required and ensure it is accessible when needed.
Expertise mapping creates systematic inventories of knowledge and capabilities distributed throughout the organization. This includes not just formal qualifications and roles, but understanding of specific process knowledge, problem-solving experience, and decision-making capabilities that may be relevant to risk assessment activities.
Dynamic expertise allocation ensures that appropriate knowledge is available for specific risk assessment challenges. This might involve bringing in experts from other sites for novel process assessments, engaging specialists for complex technical evaluations, or providing access to external expertise when internal capabilities are insufficient.
Knowledge accessibility systems make relevant information available at the point of decision-making through searchable databases, expert recommendation systems, and structured repositories that support rapid access to historical decisions, lessons learned, and validated approaches.
Knowledge Quality and Validation
Systematic assumption identification makes embedded beliefs explicit and subject to validation. Knowledge management systems must capture not just conclusions and procedures, but the assumptions and reasoning that support them. This enables systematic testing and updating when new evidence emerges.
Evidence-based knowledge validation uses operational performance data, scientific literature, and systematic observation to test the accuracy and currency of organizational knowledge. This includes both confirming successful applications and identifying instances where accepted knowledge may be incomplete or outdated.
Knowledge audit processes systematically evaluate the quality, completeness, and accessibility of knowledge required for effective risk assessment. This includes identifying knowledge gaps that may compromise assessment effectiveness and developing plans to address critical deficiencies.
Continuous knowledge improvement integrates lessons learned from risk assessment performance into organizational knowledge bases. When assessments prove accurate or identify overlooked risks, these experiences become part of organizational learning that improves future performance.
Integration with Risk Assessment Processes
Knowledge-enabled risk assessment systematically integrates relevant organizational knowledge into risk evaluation processes. This includes access to historical performance data, previous risk assessments for similar situations, lessons learned from comparable processes, and validated assumptions about process behaviors and control effectiveness.
Decision support integration provides risk assessment teams with structured access to relevant knowledge at each stage of the assessment process. This might include automated recommendations for relevant expertise, access to similar historical assessments, or prompts to consider specific knowledge domains that may be relevant.
Knowledge visualization and analytics help teams identify patterns, relationships, and insights that might not be apparent from individual data sources. This includes trend analysis, correlation identification, and systematic approaches to integrating information from multiple sources.
Real-time knowledge validation uses ongoing operational performance to continuously test and refine knowledge used in risk assessments. Rather than treating knowledge as static, these systems enable dynamic updating based on accumulating evidence and changing conditions.
A Maturity Model for Cognitive Excellence in Risk Management
Level 1: Reactive – The Bias-Blind Organization
Organizations at the reactive level operate with ad hoc risk assessments that rely heavily on individual judgment with minimal recognition of cognitive bias effects. Risk assessments are typically performed by whoever is available rather than teams with appropriate expertise, and conclusions are based primarily on immediate experience or intuitive responses.
Knowledge management characteristics at this level include isolated expertise with no systematic capture or sharing mechanisms. Critical knowledge exists primarily as tacit knowledge held by specific individuals, creating vulnerabilities when personnel changes occur. Documentation is minimal and typically focused on conclusions rather than reasoning processes or supporting evidence.
Cognitive bias manifestations are pervasive but unrecognized. Teams routinely fall prey to anchoring, confirmation bias, and availability bias without awareness of these influences on their conclusions. Unjustified assumptions are common and remain unchallenged because there are no systematic processes to identify or test them.
Decision-making processes lack structure and repeatability. Risk assessments may produce different conclusions when performed by different teams or at different times, even when addressing identical situations. There are no systematic approaches to ensuring comprehensive risk identification or validating assessment conclusions.
Typical challenges include recurring problems despite seemingly adequate risk assessments, inconsistent risk assessment quality across different teams or situations, and limited ability to learn from assessment experience. Organizations at this level often experience surprise failures where significant risks were not identified during formal risk assessment processes.
Level 2: Awareness – Recognizing the Problem
Organizations advancing to the awareness level demonstrate basic recognition of cognitive bias risks with inconsistent application of structured methods. There is growing understanding that human judgment limitations can affect risk assessment quality, but systematic approaches to addressing these limitations are incomplete or irregularly applied.
Knowledge management progress includes beginning attempts at knowledge documentation and expert identification. Organizations start to recognize the value of capturing expertise and may implement basic documentation requirements or expert directories. However, these efforts are often fragmented and lack systematic integration with risk assessment processes.
Cognitive bias recognition becomes more systematic, with training programs that help personnel understand common bias types and their potential effects on decision-making. However, awareness does not consistently translate into behavior change, and bias mitigation techniques are applied inconsistently across different assessment situations.
Decision-making improvements include basic templates or checklists that promote more systematic consideration of risk factors. However, these tools may be applied mechanically without deep understanding of their purpose or integration with broader quality system objectives.
Emerging capabilities include better documentation of assessment rationales, more systematic involvement of diverse perspectives in some assessments, and beginning recognition of the need for external expertise in complex situations. However, these practices are not yet embedded consistently throughout the organization.
Level 3: Systematic – Building Structured Defenses
Level 3 organizations implement standardized risk assessment protocols with built-in bias checks and documented decision rationales. There is systematic recognition that cognitive limitations require structured countermeasures, and processes are designed to promote more reliable decision-making.
Knowledge management formalization includes formal knowledge management processes including expert networks and structured knowledge capture. Organizations develop systematic approaches to identifying, documenting, and sharing expertise relevant to risk assessment activities. Knowledge is increasingly treated as a strategic asset requiring active management.
Bias mitigation integration embeds cognitive bias awareness and countermeasures into standard risk assessment procedures. This includes systematic use of devil’s advocate processes, structured approaches to challenging assumptions, and requirements for evidence-based justification of conclusions.
Structured decision processes ensure consistent application of comprehensive risk assessment methodologies with clear requirements for documentation, evidence, and review. Teams follow standardized approaches that promote systematic consideration of relevant risk factors while providing flexibility for situation-specific analysis.
Quality characteristics include more consistent risk assessment performance across different teams and situations, systematic documentation that enables effective review and learning, and better integration of risk assessment activities with broader quality system objectives.
Level 4: Integrated – Cultural Transformation
Level 4 organizations achieve cross-functional teams, systematic training, and continuous improvement processes with bias mitigation embedded in quality culture. Cognitive excellence becomes an organizational capability rather than a set of procedures, supported by culture, training, and systematic reinforcement.
Knowledge management integration fully integrates knowledge management with risk assessment processes and supports these with technology platforms. Knowledge flows seamlessly between different organizational functions and activities, with systematic approaches to maintaining currency and relevance of organizational knowledge assets.
Cultural integration creates organizational environments where systematic, evidence-based decision-making is expected and rewarded. Personnel at all levels understand the importance of cognitive rigor and actively support systematic approaches to risk assessment and decision-making.
Systematic training and development builds organizational capabilities in both technical risk assessment methodologies and cognitive skills required for effective application. Training programs address not just what tools to use, but how to think systematically about complex risk assessment challenges.
Continuous improvement mechanisms systematically analyze risk assessment performance to identify opportunities for enhancement and implement improvements in methodologies, training, and support systems.
Level 5: Optimizing – Predictive Intelligence
Organizations at the optimizing level implement predictive analytics, real-time bias detection, and adaptive systems that learn from assessment performance. These organizations leverage advanced technologies and systematic approaches to achieve exceptional performance in risk assessment and management.
Predictive capabilities enable organizations to anticipate potential risks and bias patterns before they manifest in assessment failures. This includes systematic monitoring of assessment performance, early warning systems for potential cognitive failures, and proactive adjustment of assessment approaches based on accumulated experience.
Adaptive learning systems continuously improve organizational capabilities based on performance feedback and changing conditions. These systems can identify emerging patterns in risk assessment challenges and automatically adjust methodologies, training programs, and support systems to maintain effectiveness.
Industry leadership characteristics include contributing to industry knowledge and best practices, serving as benchmarks for other organizations, and driving innovation in risk assessment methodologies and cognitive excellence approaches.
Implementation Strategies: Building Cognitive Excellence
Training and Development Programs
Cognitive bias awareness training must go beyond simple awareness to build practical skills in bias recognition and mitigation. Effective programs use case studies from pharmaceutical manufacturing to illustrate how biases can lead to serious consequences and provide hands-on practice with bias recognition and countermeasure application.
Critical thinking skill development builds capabilities in systematic analysis, evidence evaluation, and structured problem-solving. These programs help personnel recognize when situations require careful analysis rather than intuitive responses and provide tools for engaging systematic thinking processes.
Risk assessment methodology training combines technical instruction in formal risk assessment tools with cognitive skills required for effective application. This includes understanding when different methodologies are appropriate, how to adapt tools for specific situations, and how to recognize and address limitations in chosen approaches.
Knowledge management skills help personnel contribute effectively to organizational knowledge capture, validation, and sharing activities. This includes skills in documenting decision rationales, participating in knowledge networks, and using knowledge management systems effectively.
Technology Integration
Decision support systems provide structured frameworks that prompt systematic consideration of relevant factors while providing access to relevant organizational knowledge. These systems help teams engage appropriate cognitive processes while avoiding common bias traps.
Knowledge management platforms support effective capture, organization, and retrieval of organizational knowledge relevant to risk assessment activities. Advanced systems can provide intelligent recommendations for relevant expertise, historical assessments, and validated approaches based on assessment context.
Performance monitoring systems track risk assessment effectiveness and provide feedback for continuous improvement. These systems can identify patterns in assessment performance that suggest systematic biases or knowledge gaps requiring attention.
Collaboration tools support effective teamwork in risk assessment activities, including structured approaches to capturing diverse perspectives and managing group decision-making processes to avoid groupthink and other collective biases.
Organizational Culture Development
Leadership commitment demonstrates visible support for systematic, evidence-based approaches to risk assessment. This includes providing adequate time and resources for thorough analysis, recognizing effective risk assessment performance, and holding personnel accountable for systematic approaches to decision-making.
Psychological safety creates environments where personnel feel comfortable challenging assumptions, raising concerns about potential risks, and admitting uncertainty or knowledge limitations. This requires organizational cultures that treat questioning and systematic analysis as valuable contributions rather than obstacles to efficiency.
Learning orientation emphasizes continuous improvement in risk assessment capabilities rather than simply achieving compliance with requirements. Organizations with strong learning cultures systematically analyze assessment performance to identify improvement opportunities and implement enhancements in methodologies and capabilities.
Knowledge sharing cultures actively promote the capture and dissemination of expertise relevant to risk assessment activities. This includes recognition systems that reward knowledge sharing, systematic approaches to capturing lessons learned, and integration of knowledge management activities with performance evaluation and career development.
Conducting a Knowledge Audit for Risk Assessment
Organizations beginning this journey should start with a systematic knowledge audit that identifies potential vulnerabilities in expertise availability and access. This audit should address several key areas:
Expertise mapping to identify knowledge holders, their specific capabilities, and potential vulnerabilities from personnel changes or workload concentration. This includes both formal expertise documented in job descriptions and informal knowledge that may be critical for effective risk assessment.
Knowledge accessibility assessment to evaluate how effectively relevant knowledge can be accessed when needed for risk assessment activities. This includes both formal systems such as databases and informal networks that provide access to specialized expertise.
Knowledge quality evaluation to assess the currency, accuracy, and completeness of knowledge used to support risk assessment decisions. This includes identifying areas where assumptions may be outdated or where knowledge gaps may compromise assessment effectiveness.
Cognitive bias vulnerability assessment to identify situations where systematic biases are most likely to affect risk assessment conclusions. This includes analyzing past assessment performance to identify patterns that suggest bias effects and evaluating current processes for bias mitigation effectiveness.
Structured assessment protocols should incorporate specific checkpoints and requirements designed to counter known cognitive biases. This includes mandatory consideration of alternative explanations, requirements for external validation of conclusions, and systematic approaches to challenging preferred solutions.
Team composition guidelines should ensure appropriate cognitive diversity while maintaining technical competence. This includes balancing experience levels, functional backgrounds, and thinking styles to maximize the likelihood of identifying diverse perspectives on risk assessment challenges.
Evidence requirements should specify the types and quality of information required to support different types of risk assessment conclusions. This includes guidelines for evaluating evidence quality, addressing uncertainty, and documenting limitations in available information.
Review and validation processes should provide systematic quality checks on risk assessment conclusions while identifying potential bias effects. This includes independent review requirements, structured approaches to challenging conclusions, and systematic tracking of assessment performance over time.
Building Knowledge-Enabled Decision Making
Integration strategies should systematically connect knowledge management activities with risk assessment processes. This includes providing risk assessment teams with structured access to relevant organizational knowledge and ensuring that assessment conclusions contribute to organizational learning.
Technology selection should prioritize systems that enhance rather than replace human judgment while providing effective support for systematic decision-making processes. This includes careful evaluation of user interface design, integration with existing workflows, and alignment with organizational culture and capabilities.
Performance measurement should track both risk assessment effectiveness and knowledge management performance to ensure that both systems contribute effectively to organizational objectives. This includes metrics for knowledge quality, accessibility, and utilization as well as traditional risk assessment performance indicators.
Continuous improvement processes should systematically analyze performance in both risk assessment and knowledge management to identify enhancement opportunities and implement improvements in methodologies, training, and support systems.
Excellence Through Systematic Cognitive Development
The journey toward cognitive excellence in pharmaceutical risk management requires fundamental recognition that human cognitive limitations are not weaknesses to be overcome through training alone, but systematic realities that must be addressed through thoughtful system design. The PIC/S observations of unjustified assumptions, incomplete risk identification, and inappropriate tool application represent predictable patterns that emerge when sophisticated professionals operate without systematic support for cognitive excellence.
Excellence in this context means designing quality systems that work with human cognitive capabilities rather than against them. This requires integrating knowledge management principles with cognitive science insights to create environments where systematic, evidence-based decision-making becomes natural and sustainable. It means moving beyond hope that awareness will overcome bias toward systematic implementation of structures, processes, and cultures that promote cognitive rigor.
Elegance lies in recognizing that the most sophisticated risk assessment methodologies are only as effective as the cognitive processes that apply them. True elegance in quality system design comes from seamlessly integrating technical excellence with cognitive support, creating systems where the right decisions emerge naturally from the intersection of human expertise and systematic process.
Organizations that successfully implement these approaches will develop competitive advantages that extend far beyond regulatory compliance. They will build capabilities in systematic decision-making that improve performance across all aspects of pharmaceutical quality management. They will create resilient systems that can adapt to changing conditions while maintaining consistent effectiveness. Most importantly, they will develop cultures of excellence that attract and retain exceptional talent while continuously improving their capabilities.
The framework presented here provides a roadmap for this transformation, but each organization must adapt these principles to their specific context, culture, and capabilities. The maturity model offers a path for progressive development that builds capabilities systematically while delivering value at each stage of the journey.
As we face increasingly complex pharmaceutical manufacturing challenges and evolving regulatory expectations, the organizations that invest in systematic cognitive excellence will be best positioned to protect patient safety while achieving operational excellence. The choice is not whether to address these cognitive foundations of quality management, but how quickly and effectively we can build the capabilities required for sustained success in an increasingly demanding environment.
The cognitive foundations of pharmaceutical quality excellence represent both opportunity and imperative. The opportunity lies in developing systematic capabilities that transform good intentions into consistent results. The imperative comes from recognizing that patient safety depends not just on our technical knowledge and regulatory compliance, but on our ability to think clearly and systematically about complex risks in an uncertain world.
Reflective Questions for Implementation
How might you assess your organization’s current vulnerability to the three PIC/S observations in your risk management practices? What patterns in past risk assessment performance might indicate systematic cognitive biases affecting your decision-making processes?
Where does critical knowledge for risk assessment currently reside in your organization, and how accessible is it when decisions must be made? What knowledge audit approach would be most valuable for identifying vulnerabilities in your current risk management capabilities?
Which level of the cognitive bias mitigation maturity model best describes your organization’s current state, and what specific capabilities would be required to advance to the next level? How might you begin building these capabilities while maintaining current operational effectiveness?
What systematic changes in training, process design, and cultural expectations would be required to embed cognitive excellence into your quality culture? How would you measure progress in building these capabilities and demonstrate their value to organizational leadership?
Maintaining high-quality products is paramount, and a critical component of ensuring quality is implementing a robust review of work by a second or third person, a peer review, and/or quality review—also known as a work product review process. Like many tools, it can be underutilized. It also gets to the heart of the question of Quality Unit oversight.
Introduction to Work Product Review
Work product review systematically evaluates the output from various processes or tasks to ensure they meet predefined quality standards. This review is crucial in environments where the quality of the final product directly impacts safety and efficacy, such as in pharmaceutical manufacturing. Work product review aims to identify any deviations or defects early in the process, allowing for timely corrections and minimizing the risk of non-compliance with regulatory requirements.
Criteria for Work Product Review
To ensure that work product reviews are effective, several key criteria should be established:
Integration with Quality Management Systems: Integrate risk-based thinking into the quality management system to ensure that work product reviews are aligned with overall quality objectives. This involves regularly reviewing and updating risk assessments to reflect changes in processes or new information.
Clear Objectives: The review should have well-defined objectives that align with the process they exist within and regulatory requirements. For instance, in pharmaceutical manufacturing, these objectives might include ensuring that all documentation is accurate and complete and that manufacturing processes adhere to GMP standards.
Risk-Based: Apply work product reviews to areas identified as high-risk during the risk assessment. This ensures that resources are allocated efficiently, focusing on processes that have the greatest potential impact on quality.
Standardized Procedures: Standardized procedures should be established for conducting the review. These procedures should outline the steps involved, the reviewers’ roles and responsibilities, and the criteria for accepting or rejecting the work product.
Trained Reviewers: Reviewers should be adequately trained and competent in the subject matter. This means understanding not just the deliverable being reviewed but the regulatory framework it sits within and how it applies to the specific work products being reviewed in a GMP environment.
Documentation: All reviews should be thoroughly documented. This documentation should include the review’s results, any findings or issues identified, and actions taken to address these issues.
Feedback Loop: There should be a mechanism for feedback from the review process to improve future work products. This could involve revising procedures or providing additional training to personnel.
Bridging the Gap Between Work-as-Imagined, Work-as-Prescribed, and Work-as-Done
Work product review is a systematic process that evaluates the output from various tasks to ensure they meet predefined quality standards connecting to work-as-imagined, work-as-prescribed, and work-as-done. Work product review serves as a bridge between these concepts by systematically evaluating the output of work processes. Here’s how it connects:
Alignment with Work-as-Prescribed: Work product review ensures that outputs comply with established standards and procedures (work-as-prescribed), helping to maintain regulatory compliance and quality standards.
Insight into Work-as-Done: Through the review process, organizations gain insight into how work is actually being performed (work-as-done). This helps identify any deviations from prescribed procedures and allows for adjustments to improve alignment between work-as-prescribed and work-as-done.
Closing the Gap with Work-as-Imagined: By documenting and addressing discrepancies between work-as-imagined and work-as-done, work product review facilitates communication and feedback that can refine policies and procedures. This helps to bring work-as-imagined closer to the realities of work-as-done, improving the effectiveness of quality oversight.
Work product review is essential for ensuring that the quality of work outputs aligns with both prescribed standards and the realities of how work is actually performed. By bridging the gaps between work-as-imagined, work-as-prescribed, and work-as-done, organizations can enhance their quality management systems and maintain high standards of quality, safety and efficacy.
Aligning to the Role of Quality Unit Oversight
While work product review does not guarantee Quality Unit Oversight, it is a potential control to ensure this oversight.
In the pharmaceutical industry, the Quality Unit plays a pivotal role in ensuring drug products’ safety, efficacy, and quality. It oversees all quality-related aspects, from raw material selection to final product release. However, the Quality Unit must be enabled appropriately and structured within the organization to effectively exercise its authority and fulfill its responsibilities. This blog post explores what it means for a Quality Unit to have the necessary authority and how insufficient implementation of its responsibilities can impact pharmaceutical manufacturing.
Responsibilities of the Quality Unit
Establishing and Maintaining the Quality System: The Quality Unit must set up and continuously update the quality management system to ensure compliance with GxPs and industry best practices.
Auditing and Compliance: Conduct internal audits to ensure adherence to policies and procedures, and report quality system performance metrics.
Approving and Rejecting Components and Products: The Quality Unit has the authority to approve or reject components, drug products, and packaging materials based on quality standards.
Investigating Nonconformities: Ensuring thorough investigations into production errors, discrepancies, and complaints related to product quality.
Keeping Management Informed: Reporting on product, process, and system risks, as well as outcomes of regulatory inspections.
What It Means for a Quality Unit to Be Enabled
For a Quality Unit to be effectively enabled, it must have:
Independence: The Quality Unit should operate independently of production units to avoid conflicts of interest and ensure unbiased decision-making.
Authority: It must have the authority to approve or reject the work product without undue influence from other departments.
Resources: Adequate personnel are essential for conducting the quality unit functions.
Documentation and Procedures: Clear, documented procedures outlining responsibilities and processes are crucial for maintaining consistency and compliance.
Insufficient Implementation of Responsibilities
When a Quality Unit insufficiently implements its responsibilities, it can lead to significant issues, including:
Regulatory Noncompliance: Failure to adhere to GxPs and regulatory standards can result in regulatory action.
Product Quality Issues: Inadequate oversight can lead to the release of substandard products, posing risks to patient safety and public health.
Lack of Continuous Improvement: Without effective quality systems in place, opportunities for process improvements and innovation may be missed.
The Quality Unit is the backbone of pharmaceutical manufacturing, ensuring that products meet the highest standards of quality and safety. By understanding the Quality Unit’s responsibilities and ensuring it has the necessary authority and resources, pharmaceutical companies can maintain compliance, protect public health, and foster a culture of continuous improvement. Inadequate implementation of these responsibilities can have severe consequences, emphasizing the importance of a well-structured and empowered Quality Unit.
By understanding these responsibilities, we can take a risk-based approach to applying quality review.
When to Apply Quality Review as Work Product Review
Work product review by Quality should be applied at critical stages to guarantee critical-to-quality attributes, including adherence to the regulations. This should be a risk-based approach. As such, it should be identified as controls in a living risks assessment and adjusted (add more, remove where unnecessary) as appropriate.
Closely scrutinize the responsibilities of the Quality Unit in the regulations to ensure all are met.
Rubrics are a great way to standardize quality reviews. If it is important enough to require a work review, it is important enough to standardize. The process owner should develop and maintain these rubrics with an appropriate group of stakeholder custodians. This is a key part of knowledge management. Having this cross-functional perspective on the output and what quality looks like is critical. This rubric should include:
Definition of prescribed work and the intended output that is being reviewed
Potential outcomes related to critical attributes, including definitions of technical accuracy
Methods and techniques used to generate the outcome
Operating experience and lessons learned
Risks, hazards, and user-centered design considerations
Requirements, standards, and code compliance
Planning, oversight, and acceptance testing
Input data and sources
Assumptions
Documentation required
Reviews and approvals required
Program or procedural obstacles to desired performance
Surprise situations, for example, unanticipated risk factors, schedule or scope changes, and organizational issues
Engineering human performance tool(s) applicable to activities being reviewed.
The rubric should have an assessment component, and that assessment should feed back into the originator’s qualified state.
Work product reviews must be early enough to allow feedback into the normal work for repetitive tasks. This should lead to gates in processes, quality-on-the-floor, or better-trained supervisors performing better and more effective reviews. This feedback should always be to the responsible person – the originator—and should be, wherever possible, face-to-face feedback to resolve the particular issues identified. This dialogue is critical.
Conclusion
Work product review is a powerful tool for enhancing quality oversight. By aligning this process with the responsibilities of the Quality Unit and implementing best practices such as standardized rubrics and a risk-based approach, companies can ensure that their products meet the highest standards of quality and safety. Effective work product review not only supports regulatory compliance but also fosters a culture of continuous improvement, which is essential for maintaining excellence in the pharmaceutical industry.
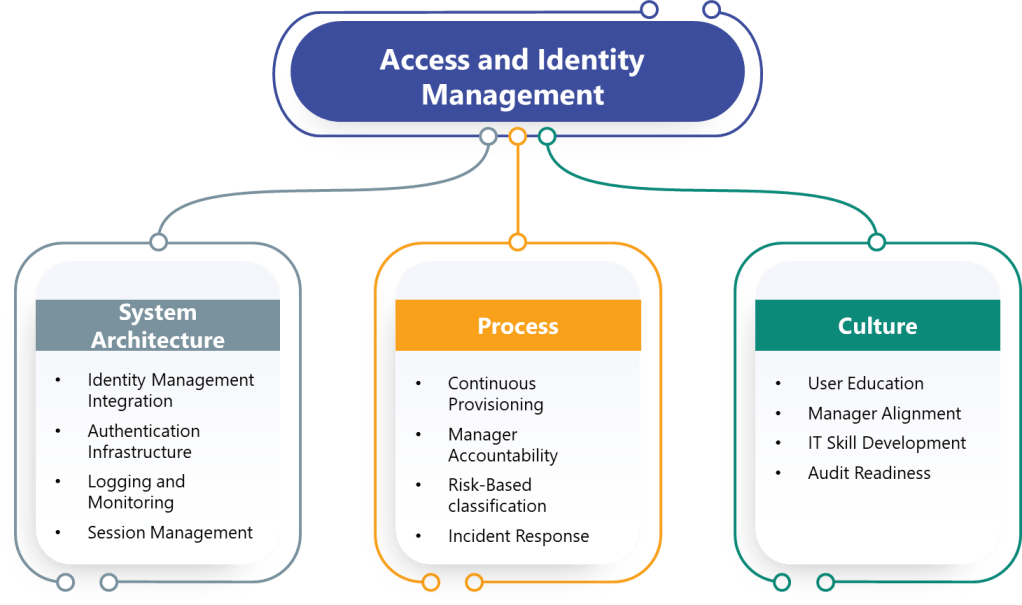
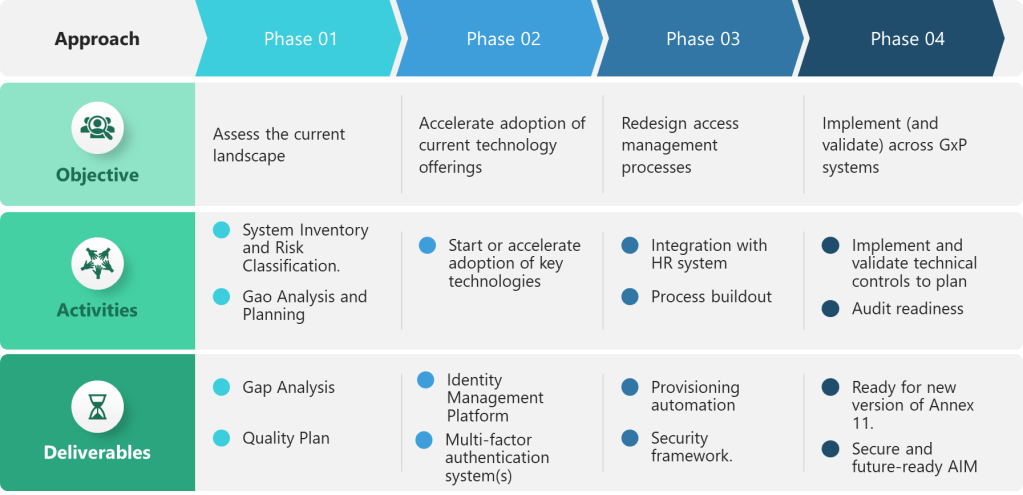
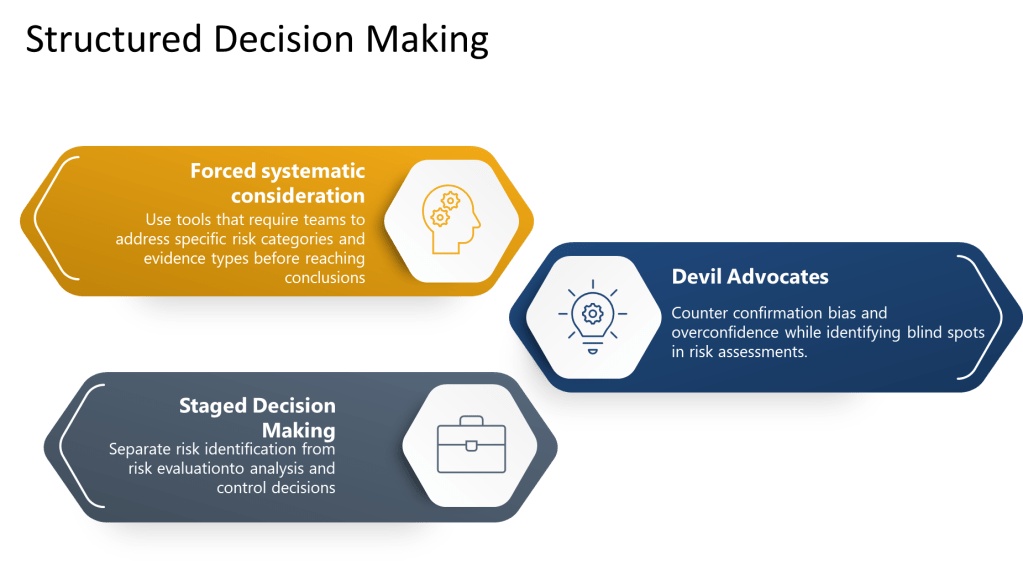
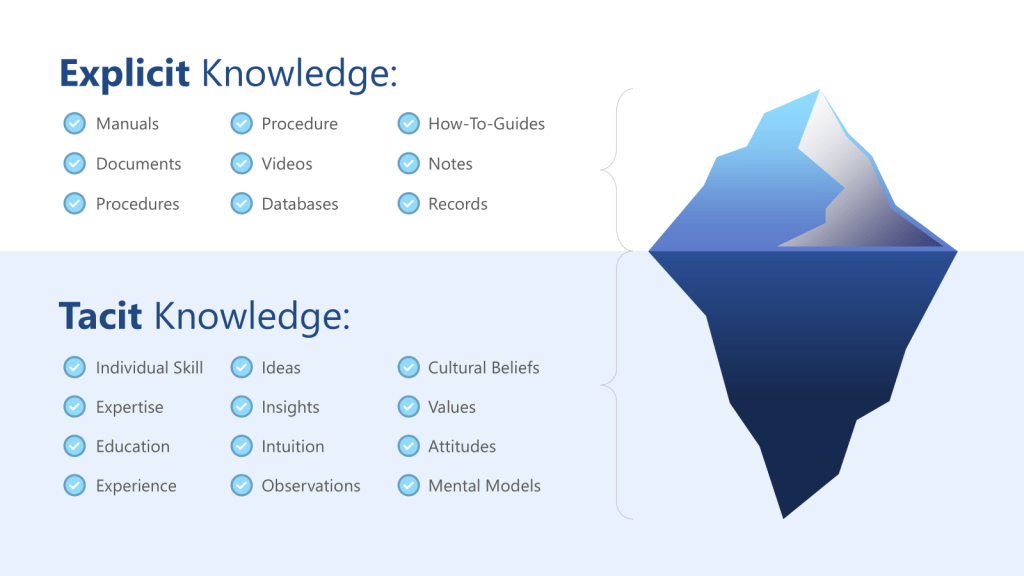
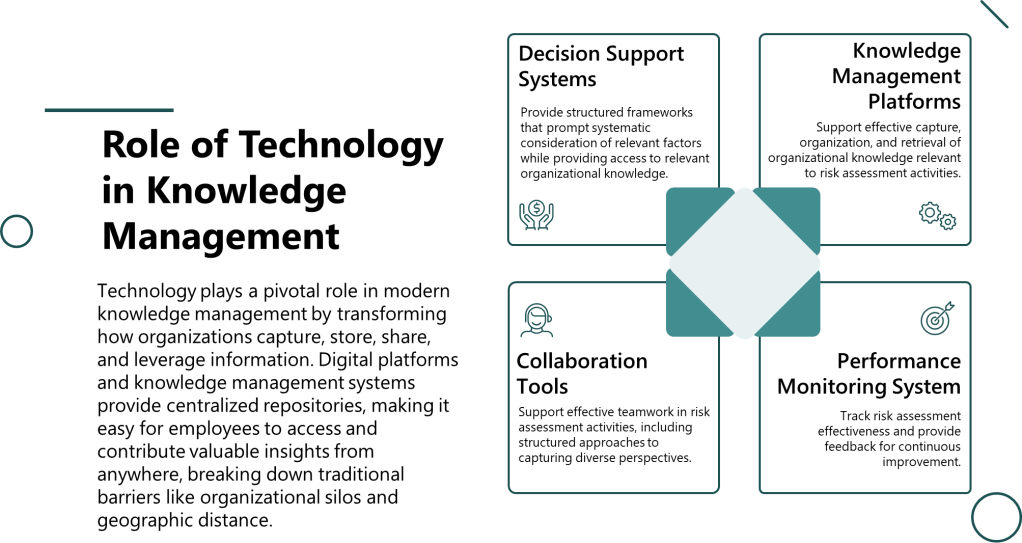


{kind=link}