The draft revision of Eudralex Volume 4 Chapter 1 marks a substantial evolution from the current version, reflecting regulatory alignment with ICH Q9(R1), enhanced risk-based approaches, and a new emphasis on knowledge management, proactive risk detection, and supply chain resilience.
Core Differences at a Glance
The draft update integrates advances in global quality science—especially from ICH Q9(R1)—anchoring the Pharmaceutical Quality System (PQS) more firmly in knowledge management and risk management practice.
Proactive risk identification and mitigation are highlighted, reflecting the need to anticipate supply disruptions and quality failures, beyond routine compliance.
The requirements for Product Quality Review (PQR) are clarified, notably in how to handle grouped products and limited-batch scenarios, enhancing operational clarity for diverse manufacturing models.
Philosophical Shift: From Compliance to Dynamic Risk Management
Where the current Chapter 1 (in force since 2013) framed the PQS largely as a static structure of roles, documentation, and reviews, the draft version pivots toward a learning organization approach: knowledge acquisition, use, and feedback become core system elements.
Emphasis is now placed on systematic knowledge management as both a regulatory and operational priority. This serves as an overt marker of quality system maturity, intended to reduce “invisible failures” and foster analytical vigilance—aligning closely with falsifiable quality frameworks.
Risk-Based Decision-Making: Explicit and Actionable
The revision operationalizes risk-based thinking by mandating scientific rationale for risk decisions and clarifying expectations for proportionality in risk assessment. The regulator’s intent is clear: risk management can no longer be a box-checking exercise, but must be demonstrably linked to daily site operations and lifecycle decisions.
This brings the PQS into closer alignment with both the adaptive toolbox and the take-the-best heuristics: decisive focus on the most causally relevant risk vectors rather than exhaustive factor listing, echoing playbooks for effective investigation and CAPA prioritization.
Product Quality Review (PQR) and Batch Grouping
Clarification is provided in the revised text on how to perform quality reviews for products manufactured in small numbers or as grouped products, a challenge long met with uncertainty. The draft provides operational guidance, aiming to resolve ambiguities around the statistical and process review requirements for product families and low-volume production.
Supply Chain Resilience, Shortage Prevention, and Knowledge Networks
The draft gives unprecedented attention to shortage prevention and supply chain risk. Manufacturers will be expected to anticipate, document, and mitigate vulnerabilities not only in routine operations but also in emergency or shortage-prone contexts. This aligns the PQS with broader public health objectives, situating quality management as a bulwark against systemic healthcare risk.
International Harmonization and the ICH Q9(R1) Impact
Most significantly, the update explicitly references alignment with ICH Q9(R1) on Quality Risk Management, making harmonization with international best practice an explicit goal. This pushes organizations toward the global baseline for science- and risk-driven GMP.
The effect will be increased regulatory predictability for multinational manufacturers and heightened expectations for knowledge-handling and continuous improvement.
Summary Table: Draft vs. Current Chapter 1
Feature
Current Chapter 1 (2013)
Draft Chapter 1 (2025)
PQS Philosophy
Compliance/document control
Knowledge management & risk management
Risk Management
Implied, periodic
Embedded, real-time, evidence-based
ICH Q9 Alignment
Partial
Explicit, full alignment to Q9(R1)
Product Quality Review (PQR)
General guidance
Detailed, incl. grouped/low-batch
Supply Chain & Shortages
Minimal focus
Proactive risk, shortage prevention
Corrective/Preventive Action (CAPA)
System-oriented
Rooted in risk, causal prioritization
Lifecycle Integration
Weak
Strong, with embedded feedback
Operational Implications for Quality Leaders
The new Chapter 1 will demand a more dynamic, evidence-driven PQS, with robust mechanisms for knowledge transfer, risk-based priority setting, and system learning cycles. Technical writing, investigation reports, and CAPA logic will need to reference causal mechanisms and risk rationale explicitly—a marked shift from checklists to analytical narratives, aligning with the take-the-best causal reasoning discussed in your recent writings.
To prepare, organizations should:
Review and strengthen knowledge management assets
Embed risk assessment into the daily decision matrix—not just annual reviews
Foster investigative cultures that value causal specificity over exhaustive documentation
Reframe supply chain oversight as a continuous risk monitoring exercise
This systemic move, when enacted, will shift GMP thinking from historical compliance to forward-looking, adaptive quality management—an ambitious but necessary corrective for the challenges facing pharmaceutical manufacturing in 2025 and beyond.
The draft revision of EU GMP Chapter 4 introduces what can only be described as a revolutionary framework for data governance systems. This isn’t merely an update to existing documentation requirements—it is a keystone document that cements the decade long paradigm shift of data governance as the cornerstone of modern pharmaceutical quality systems.
The Genesis of Systematic Data Governance
The most striking aspect of the draft Chapter 4 is the introduction of sections 4.10 through 4.18, which establish data governance systems as mandatory infrastructure within pharmaceutical quality systems. This comprehensive framework emerges from lessons learned during the past decade of data integrity enforcement actions and reflects the reality that modern pharmaceutical manufacturing operates in an increasingly digital environment where traditional documentation approaches are insufficient.
The requirement that regulated users “establish a data governance system integral to the pharmaceutical quality system” moves far beyond the current Chapter 4’s basic documentation requirements. This integration ensures that data governance isn’t treated as an IT afterthought or compliance checkbox, but rather as a fundamental component of how pharmaceutical companies ensure product quality and patient safety. The emphasis on integration with existing pharmaceutical quality systems builds on synergies that I’ve previously discussed in my analysis of how data governance, data quality, and data integrity work together as interconnected pillars.
The requirement for regular documentation and review of data governance arrangements establishes accountability and ensures continuous improvement. This aligns with my observations about risk-based thinking where effective quality systems must anticipate, monitor, respond, and learn from their operational environment.
Comprehensive Data Lifecycle Management
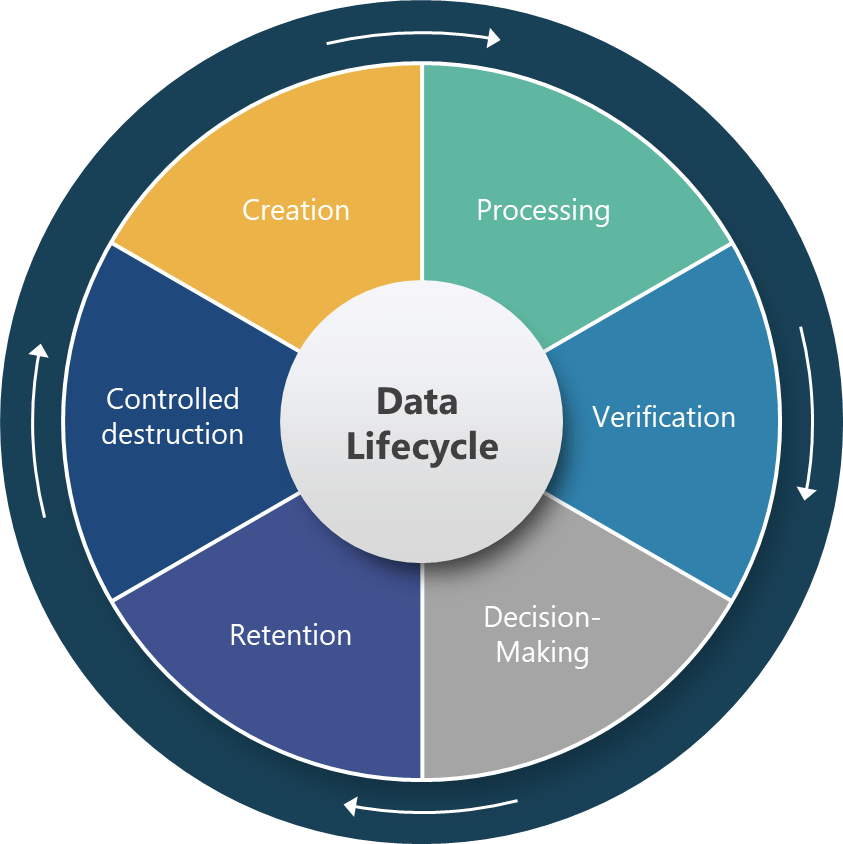
Section 4.12 represents perhaps the most technically sophisticated requirement in the draft, establishing a six-stage data lifecycle framework that covers creation, processing, verification, decision-making, retention, and controlled destruction. This approach acknowledges that data integrity cannot be ensured through point-in-time controls but requires systematic management throughout the entire data journey.
The specific requirement for “reconstruction of all data processing activities” for derived data establishes unprecedented expectations for data traceability and transparency. This requirement will fundamentally change how pharmaceutical companies design their data processing workflows, particularly in areas like process analytical technology (PAT), manufacturing execution systems (MES), and automated batch release systems where raw data undergoes significant transformation before supporting critical quality decisions.
The lifecycle approach also creates direct connections to computerized system validation requirements under Annex 11, as noted in section 4.22. This integration ensures that data governance systems are not separate from, but deeply integrated with, the technical systems that create, process, and store pharmaceutical data. As I’ve discussed in my analysis of computer system validation frameworks, effective validation programs must consider the entire system ecosystem, not just individual software applications.
Risk-Based Data Criticality Assessment
The draft introduces a sophisticated two-dimensional risk assessment framework through section 4.13, requiring organizations to evaluate both data criticality and data risk. Data criticality focuses on the impact to decision-making and product quality, while data risk considers the opportunity for alteration or deletion and the likelihood of detection. This framework provides a scientific basis for prioritizing data protection efforts and designing appropriate controls.
This approach represents a significant evolution from current practices where data integrity controls are often applied uniformly regardless of the actual risk or impact of specific data elements. The risk-based framework allows organizations to focus their most intensive controls on the data that matters most while applying appropriate but proportionate controls to lower-risk information. This aligns with principles I’ve discussed regarding quality risk management under ICH Q9(R1), where structured, science-based approaches reduce subjectivity and improve decision-making.
The requirement to assess “likelihood of detection” introduces a crucial element often missing from traditional data integrity approaches. Organizations must evaluate not only how to prevent data integrity failures but also how quickly and reliably they can detect failures that occur despite preventive controls. This assessment drives requirements for monitoring systems, audit trail analysis capabilities, and incident detection procedures.
Service Provider Oversight and Accountability
Section 4.18 establishes specific requirements for overseeing service providers’ data management policies and risk control strategies. This requirement acknowledges the reality that modern pharmaceutical operations depend heavily on cloud services, SaaS platforms, contract manufacturing organizations, and other external providers whose data management practices directly impact pharmaceutical company compliance.
The risk-based frequency requirement for service provider reviews represents a practical approach that allows organizations to focus oversight efforts where they matter most while ensuring that all service providers receive appropriate attention. For more details on the evolving regulatory expectations around supplier management see the post “draft Annex 11’s supplier oversight requirements“.
The service provider oversight requirement also creates accountability throughout the pharmaceutical supply chain, ensuring that data integrity expectations extend beyond the pharmaceutical company’s direct operations to encompass all entities that handle GMP-relevant data. This approach recognizes that regulatory accountability cannot be transferred to external providers, even when specific activities are outsourced.
Operational Implementation Challenges
The transition to mandatory data governance systems will present significant operational challenges for most pharmaceutical organizations. The requirement for “suitably designed systems, the use of technologies and data security measures, combined with specific expertise” in section 4.14 acknowledges that effective data governance requires both technological infrastructure and human expertise.
Organizations will need to invest in personnel with specialized data governance expertise, implement technology systems capable of supporting comprehensive data lifecycle management, and develop procedures for managing the complex interactions between data governance requirements and existing quality systems. This represents a substantial change management challenge that will require executive commitment and cross-functional collaboration.
The requirement for regular review of risk mitigation effectiveness in section 4.17 establishes data governance as a continuous improvement discipline rather than a one-time implementation project. Organizations must develop capabilities for monitoring the performance of their data governance systems and adjusting controls as risks evolve or new technologies are implemented.
The integration with quality risk management principles throughout sections 4.10-4.22 creates powerful synergies between traditional pharmaceutical quality systems and modern data management practices. This integration ensures that data governance supports rather than competes with existing quality initiatives while providing a systematic framework for managing the increasing complexity of pharmaceutical data environments.
The draft’s emphasis on data ownership throughout the lifecycle in section 4.15 establishes clear accountability that will help organizations avoid the diffusion of responsibility that often undermines data integrity initiatives. Clear ownership models provide the foundation for effective governance, accountability, and continuous improvement.
The draft revision of EU GMP Chapter 4 on Documentation represents more than just an update—it signals a paradigm shift toward digitalization, enhanced data integrity, and risk-based quality management in pharmaceutical manufacturing.
The Digital Transformation Imperative
The draft Chapter 4 emerges from a recognition that pharmaceutical manufacturing has fundamentally changed since 2011. The rise of Industry 4.0, artificial intelligence in manufacturing decisions, and the critical importance of data integrity following numerous regulatory actions have necessitated a complete reconceptualization of documentation requirements.
The new framework introduces comprehensive data governance systems, risk-based approaches throughout the documentation lifecycle, and explicit requirements for hybrid systems that combine paper and electronic elements. These changes reflect lessons learned from data integrity violations that have cost the industry billions in remediation and lost revenue.
Detailed Document Type Analysis
Master Documents: Foundation of Quality Systems
Document Type
Current Chapter 4 (2011) Requirements
Draft Chapter 4 (2025) Requirements
FDA 21 CFR 211
ICH Q7
WHO GMP
ISO 13485
Site Master File
A document describing the GMP related activities of the manufacturer
Refer to EU GMP Guidelines, Volume 4 ‘Explanatory Notes on the preparation of a Site Master File’
No specific equivalent, but facility information requirements under §211.176
Section 2.5 – Documentation system should include site master file equivalent information
Section 4.1 – Site master file requirements similar to EU GMP
Quality manual requirements under Section 4.2.2
Validation Master Plan
Not specified
A document describing the key elements of the site qualification and validation program
Process validation requirements under §211.100 and §211.110
Section 12 – Validation requirements for critical operations
Section 4.2 – Validation and qualification programs
Validation planning under Section 7.5.6 and design validation
The introduction of the Validation Master Plan as a mandatory master document represents the most significant addition to this category. This change acknowledges the critical role of systematic validation in modern pharmaceutical manufacturing and aligns EU GMP with global best practices seen in FDA and ICH frameworks.
The Site Master File requirement, while maintained, now references more detailed guidance, suggesting increased regulatory scrutiny of facility information and manufacturing capabilities.
Instructions: The Operational Backbone
Document Type
Current Chapter 4 (2011) Requirements
Draft Chapter 4 (2025) Requirements
FDA 21 CFR 211
ICH Q7
WHO GMP
ISO 13485
Specifications
Describe in detail the requirements with which the products or materials used or obtained during manufacture have to conform. They serve as a basis for quality evaluation
Refer to glossary for definition
Component specifications §211.84, drug product specifications §211.160
Section 7.3 – Specifications for starting materials, intermediates, and APIs
Section 4.12 – Specifications for starting materials and finished products
Requirements specifications under Section 7.2.1
Manufacturing Formulae, Processing, Packaging and Testing Instructions
Provide detail all the starting materials, equipment and computerised systems (if any) to be used and specify all processing, packaging, sampling and testing instructions
Provide complete detail on all the starting materials, equipment, and computerised systems (if any) to be used and specify all processing, packaging, sampling, and testing instructions to ensure batch to batch consistency
Master production and control records §211.186, production record requirements §211.188
Section 6.4 – Master production instructions and batch production records
Section 4.13 – Manufacturing formulae and processing instructions
Production and service provision instructions Section 7.5.1
Procedures (SOPs)
Give directions for performing certain operations
Otherwise known as Standard Operating Procedures, documented set of instructions for performing and recording operations
Written procedures required throughout Part 211 for various operations
Section 6.1 – Written procedures for all critical operations
Section 4.14 – Standard operating procedures for all operations
Documented procedures throughout the standard, Section 4.2.1
Technical/Quality Agreements
Are agreed between contract givers and acceptors for outsourced activities
Written proof of agreement between contract givers and acceptors for outsourced activities
Section 16 – Contract manufacturers agreements and responsibilities
Section 7 – Contract manufacture and analysis agreements
Outsourcing agreements under Section 7.4 – Purchasing
The enhancement of Manufacturing Instructions to explicitly require “batch to batch consistency” represents a crucial evolution. This change reflects increased regulatory focus on manufacturing reproducibility and aligns with FDA’s process validation lifecycle approach and ICH Q7’s emphasis on consistent API production.
Procedures (SOPs) now explicitly encompass both “performing and recording operations,” emphasizing the dual nature of documentation as both instruction and evidence creation1. This mirrors FDA 21 CFR 211’s comprehensive procedural requirements and ISO 13485’s systematic approach to documented procedures910.
The transformation of Technical Agreements into Technical/Quality Agreements with emphasis on “written proof” reflects lessons learned from outsourcing challenges and regulatory enforcement actions. This change aligns with ICH Q7’s detailed contract manufacturer requirements and strengthens oversight of critical outsourced activities.
Records and Reports: Evidence of Compliance
Document Type
Current Chapter 4 (2011) Requirements
Draft Chapter 4 (2025) Requirements
FDA 21 CFR 211
ICH Q7
WHO GMP
ISO 13485
Records
Provide evidence of various actions taken to demonstrate compliance with instructions, e.g. activities, events, investigations, and in the case of manufactured batches a history of each batch of product
Provide evidence of various actions taken to demonstrate compliance with instructions, e.g. activities, events, investigations, and in the case of manufactured batches a history of each batch of product, including its distribution. Records include the raw data which is used to generate other records
Comprehensive record requirements throughout Part 211, §211.180 general requirements
Section 6.5 – Batch production records and Section 6.6 – Laboratory control records
Section 4.16 – Records requirements for all GMP activities
Quality records requirements under Section 4.2.4
Certificate of Analysis
Provide a summary of testing results on samples of products or materials together with the evaluation for compliance to a stated specification
Provide a summary of testing results on samples of products or materials together with the evaluation for compliance to a stated specification
Laboratory records and test results §211.194, certificate requirements
Section 11.15 – Certificate of analysis for APIs
Section 6.8 – Certificates of analysis requirements
Test records and certificates under Section 7.5.3
Reports
Document the conduct of particular exercises, projects or investigations, together with results, conclusions and recommendations
Document the conduct of exercises, studies, assessments, projects or investigations, together with results, conclusions and recommendations
The expansion of Recordsto explicitly include “raw data” and “distribution information” represents perhaps the most impactful change for day-to-day operations. This enhancement directly addresses data integrity concerns highlighted by regulatory inspections and enforcement actions globally. The definition now states that “Records include the raw data which is used to generate other records,” establishing clear expectations for data traceability that align with FDA’s data integrity guidance and ICH Q7’s comprehensive record requirements.
Reports now encompass “exercises, studies, assessments, projects or investigations,” broadening the scope beyond the current “particular exercises, projects or investigations”. This expansion aligns with modern pharmaceutical operations that increasingly rely on various analytical studies and assessments for decision-making, matching ISO 13485’s comprehensive reporting requirements.
Revolutionary Framework Elements
Data Governance Revolution
The draft introduces an entirely new paradigm through its Data Governance Systems (Sections 4.10-4.18). This framework establishes:
Complete lifecycle management from data creation through retirement
Risk-based approaches considering data criticality and data risk
Service provider oversight with periodic review requirements
Ownership accountability throughout the data lifecycle
This comprehensive approach exceeds traditional GMP requirements and positions EU regulations at the forefront of data integrity management, surpassing even FDA’s current frameworks in systematic approach.
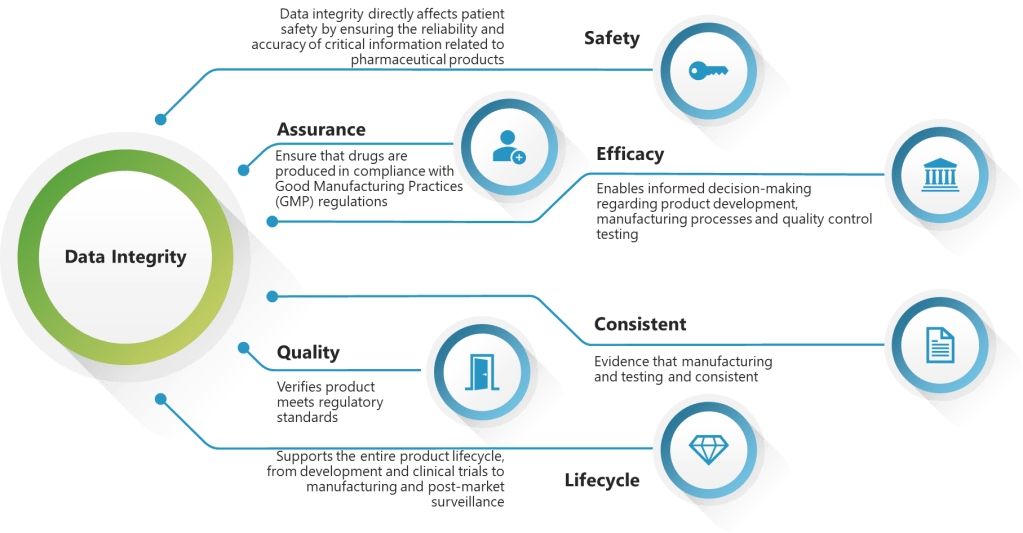
ALCOA++ Formalization
The draft formalizes ALCOA++ principles (Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, Available, Traceable) with detailed definitions for each attribute. This represents a major comprehensive regulatory codification of these principles, providing unprecedented clarity for industry implementation.
ALCOA++ Principles: Comprehensive Data Integrity Framework
The Draft EU GMP Chapter 4 (2025) formalizes the ALCOA++ principles as the foundation for data integrity in pharmaceutical manufacturing. This represents the first comprehensive regulatory codification of these expanded data integrity principles, building upon the traditional ALCOA framework with five additional critical elements.
Complete ALCOA++ Requirements Table
Principle
Core Requirement
Paper Implementation
Electronic Implementation
A – Attributable
Identify who performed the task and when
Signatures, dates, initials
User authentication, e-signatures
L – Legible
Information must be readable and unambiguous
Clear writing, permanent ink
Proper formats, search functionality
C – Contemporaneous
Record actions as they happen in real-time
Immediate recording
System timestamps, workflow controls
O – Original
Preserve first capture of information
Original documents retained
Database integrity, backups
A – Accurate
Ensure truthful representation of facts
Training, calibrated equipment
System validation, automated checks
+ Complete
Include all critical information and metadata
Complete data, no missing pages
Metadata capture, completeness checks
+ Consistent
Standardize data creation and processing
Standard formats, consistent units
Data standards, validation rules
+ Enduring
Maintain records throughout retention period
Archival materials, proper storage
Database integrity, migration plans
+ Available
Ensure accessibility for authorized personnel
Organized filing, access controls
Role-based access, query capabilities
+ Traceable
Enable tracing of data history and changes
Sequential numbering, change logs
Audit trails, version control
Hybrid Systems Management
Recognizing the reality of modern pharmaceutical operations, the draft dedicates sections 4.82-4.85 to hybrid systems that combine paper and electronic elements. This practical approach acknowledges that many manufacturers operate in mixed environments and provides specific requirements for managing these complex systems.
A New Era of Pharmaceutical Documentation
The draft EU GMP Chapter 4 represents the most significant evolution in pharmaceutical documentation requirements in over a decade. By introducing comprehensive data governance frameworks, formalizing data integrity principles, and acknowledging the reality of digital transformation, these changes position European regulations as global leaders in modern pharmaceutical quality management.
For industry professionals, these changes offer both challenges and opportunities. Organizations that proactively embrace these new paradigms will not only achieve regulatory compliance but will also realize operational benefits through improved data quality, enhanced decision-making capabilities, and reduced compliance costs.
The evolution from simple documentation requirements to comprehensive data governance systems reflects the maturation of the pharmaceutical industry and its embrace of digital technologies. As we move toward implementation, the industry’s response to these changes will shape the future of pharmaceutical manufacturing for decades to come.
The message is clear: the future of pharmaceutical documentation is digital, risk-based, and comprehensive. Organizations that recognize this shift and act accordingly will thrive in the new regulatory environment, while those that cling to outdated approaches risk being left behind in an increasingly sophisticated and demanding regulatory landscape.
The environment for commissioning, qualification, and validation (CQV) professionals remains defined by persistent challenges. Rapid technological advancements—most notably in artificial intelligence, machine learning, and automation—are constantly reshaping the expectations for validation. Compliance requirements are in frequent flux as agencies modernize guidance, while the complexity of novel biologics and therapies demands ever-higher standards of sterility, traceability, and process control. The shift towards digital systems has introduced significant hurdles in data management and integration, often stretching already limited resources. At the same time, organizations are expected to fully embrace risk-based, science-first approaches, which require new methodologies and skills. Finally, true validation now hinges on effective collaboration and knowledge-sharing among increasingly cross-functional and global teams.
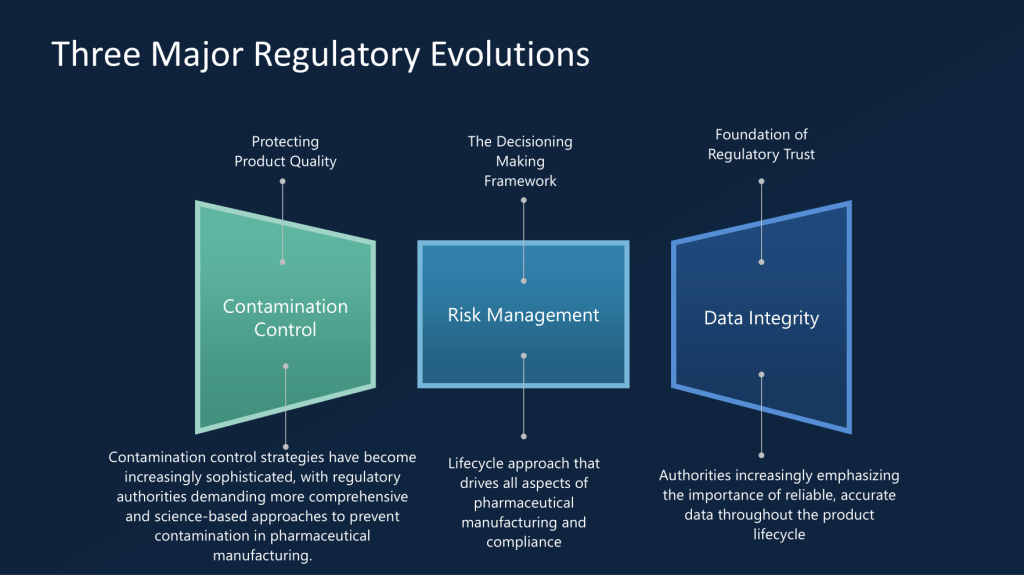
Overlaying these challenges, three major regulatory paradigm shifts are transforming the expectations around risk management, contamination control, and data integrity. Data integrity in particular has become an international touchpoint. Since the landmark PIC/S guidance in 2021 and matching World Health Organization updates, agencies have made it clear that trustworthy, accurate, and defendable data—whether paper-based or digital—are the foundation of regulatory confidence. Comprehensive data governance, end-to-end traceability, and robust documentation are now all non-negotiable.
Contamination control is experiencing its own revolution. The August 2023 overhaul of EU GMP Annex 1 set a new benchmark for sterile manufacturing. The core concept, the Contamination Control Strategy (CCS), formalizes expectations: every manufacturer must systematically identify, map, and control contamination risks across the entire product lifecycle. From supply chain vigilance to environmental monitoring, regulators are pushing for a proactive, science-driven, and holistic approach, far beyond previous practices that too often relied on reactive measures. We this reflected in recent USP drafts as well.
Quality risk management (QRM) also has a new regulatory backbone. The ICH Q9(R1) revision, finalized in 2023, addresses long-standing shortcomings—particularly subjectivity and lack of consistency—in how risks are identified and managed. The European Medicines Agency’s ongoing revision of EudraLex Chapter 1, now aiming for finalization in 2026, will further require organizations to embed preventative, science-based risk management within globalized and complex supply chain operations. Modern products and supply webs simply cannot be managed with last-generation compliance thinking.
The EU Digital Modernization: Chapter 4, Annex 11, and Annex 22
With the rapid digitalization of pharma, the European Union has embarked on an ambitious modernization of its GMP framework. At the heart of these changes are the upcoming revisions to Chapter 4 (Documentation), Annex 11 (Computerised Systems), and the anticipated implementation of Annex 22 (Artificial Intelligence).
Chapter 4—Documentation is being thoroughly updated in parallel with Annex 11. The current chapter, which governs all aspects of documentation in GMP environments, was last revised in 2011. Its modernization is a direct response to the prevalence of digital tools—electronic records, digital signatures, and interconnected documentation systems. The revised Chapter 4 is expected to provide much clearer requirements for the management, review, retention, and security of both paper and electronic records, ensuring that information flows align seamlessly with the increasingly digital processes described in Annex 11. Together, these updates will enable companies to phase out paper where possible, provided electronic systems are validated, auditable, and secure.
Annex 11—Computerised Systems will see its most significant overhaul since the dawn of digital pharma. The new guidance, scheduled for publication and adoption in 2026, directly addresses areas that the previous version left insufficiently covered. The scope now embraces the tectonic shift toward AI, machine learning, cloud-based services, agile project management, and advanced digital workflows. For instance, close attention is being paid to the robustness of electronic signatures, demanding multi-factor authentication, time-zoned audit trails, and explicit provisions for non-repudiation. Hybrid (wet-ink/digital) records will only be acceptable if they can demonstrate tamper-evidence via hashes or equivalent mechanisms. Especially significant is the regulation of “open systems” such as SaaS and cloud platforms. Here, organizations can no longer rely on traditional username/password models; instead, compliance with standards like eIDAS for trusted digital providers is expected, with more of the technical compliance burden shifting onto certified digital partners.
The new Annex 11 also calls for enhanced technical controls throughout computerized systems, proportional risk management protocols for new technologies, and a far greater emphasis on continuous supplier oversight and lifecycle validation. Integration with the revised Chapter 4 ensures that documentation requirements and data management are harmonized across the digital value chain.
The introduction of Annex 22 represents a pivotal moment in the regulatory landscape for pharmaceutical manufacturing in Europe. This annex is the EU’s first dedicated framework addressing the use of Artificial Intelligence (AI) and machine learning in the production of active substances and medicinal products, responding to the rapid digital transformation now reshaping the industry.
Annex 22 sets out explicit requirements to ensure that any AI-based systems integrated into GMP-regulated environments are rigorously controlled and demonstrably trustworthy. It starts by mandating that manufacturers clearly define the intended use of any AI model deployed, ensuring its purpose is scientifically justified and risk-appropriate.
Quality risk management forms the backbone of Annex 22. Manufacturers must establish performance metrics tailored to the specific application and product risk profile of AI, and they are required to demonstrate the suitability and adequacy of all data used for model training, validation, and testing. Strong data governance principles apply: manufacturers need robust controls over data quality, traceability, and security throughout the AI system’s lifecycle.
The annex foresees a continuous oversight regime. This includes change control processes for AI models, ongoing monitoring of performance to detect drift or failures, and formally documented procedures for human intervention where necessary. The emphasis is on ensuring that, even as AI augments or automates manufacturing processes, human review and responsibility remain central for all quality- and safety-critical steps.
By introducing these requirements, Annex 22 aims to provide sufficient flexibility to enable innovation, while anchoring AI applications within a robust regulatory framework that safeguards product quality and patient safety at every stage. Together with the updates to Chapter 4 and Annex 11, Annex 22 gives companies clear, actionable expectations for responsibly harnessing digital innovation in the manufacturing environment.
Life Cycle Integration, Analytical Validation, and AI/ML Guidance
Across global regulators, a clear consensus has taken shape: validation must be seen as a continuous lifecycle process, not as a “check-the-box” activity. The latest WHO technical reports, the USP’s evolving chapters (notably <1058> and <1220>), and the harmonized ICH Q14 all signal a new age of ongoing qualification, continuous assurance, change management, and systematic performance verification. The scope of validation stretches from the design qualification stage through annual review and revalidation after every significant change.
A parallel wave of guidance for AI and machine learning is cresting. The EMA, FDA, MHRA, and WHO are now releasing coordinated documents addressing everything from transparent model architecture and dataset controls to rigorous “human-in-the-loop” safeguards for critical manufacturing decisions, including the new draft Annex 22. Data governance—traceability, security, and data quality—has never been under more scrutiny.
Regulatory Body
Document Title
Publication Date
Status
Key Focus Areas
EMA
Reflection Paper on the Use of Artificial Intelligence in the Medicinal Product Lifecycle
Oct-24
Final
Risk-based approach for AI/ML development, deployment, and performance monitoring across product lifecycle including manufacturing
EMA/HMA
Multi-annual AI Workplan 2023-2028
Dec-23
Final
Strategic framework for European medicines regulatory network to utilize AI while managing risks
EMA
Annex 22 Artificial Intelligence
Jul-25
Draft
Establishes requirements for the use of AI and machine learning in the manufacturing of active substances and medicinal products.
FDA
Considerations for the Use of AI to Support Regulatory Decision Making for Drug and Biological Products
Feb-25
Draft
Guidelines for using AI to generate information for regulatory submissions
FDA
Discussion Paper on AI in the Manufacture of Medicines
May-23
Published
Considerations for cloud applications, IoT data management, regulatory oversight of AI in manufacturing
FDA/Health Canada/MHRA
Good Machine Learning Practice for Medical Device Development Guiding Principles
Mar-25
Final
10 principles to inform development of Good Machine Learning Practice
WHO
Guidelines for AI Regulation in Health Care
Oct-23
Final
Six regulatory areas including transparency, risk management, data quality
MHRA
AI Regulatory Strategy
Apr-24
Final
Strategic approach based on safety, transparency, fairness, accountability, and contestability principles
EFPIA
Position Paper on Application of AI in a GMP Manufacturing Environment
Sep-24
Published
Industry position on using existing GMP framework to embrace AI/ML solutions
The Time is Now
The world of validation is no longer controlled by periodic updates or leisurely transitions. Change is the new baseline. Regulatory authorities have codified the digital, risk-based, and globally harmonized future—are your systems, people, and partners ready?