This year’s rant is triggered by reading a good practices guide designed to be pan-GxP and getting frustrated by its utter GMP focus. I knew I was in trouble when it specifically discussed “Product and Process Understanding” as a critical factor and then referenced ICH Q10. Use those terms with ICH Q10, and you just announced to the entire world that this is a GMP book. It is important to use a wider term and then reference product/process understanding as one subcategory or way of meeting it.
I rather like the approach of ICH E6 and E8 here, which is to use the wider term “Critical to Quality,” which in the broader sense can be expanded to mean the key factors that must be controlled or monitored to ensure the quality, safety, and efficacy of pharmaceutical products from development to clinical studies to manufacturing and distribution and beyond. It’s a risk-based approach focused on what matters most for patient safety and reliable results.
There has been increasing evidence in recent years that research in life sciences is lacking in reproducibility and data quality. This raises the need for effective systems to improve data integrity in the evolving non-GxP research environment. Reproducibility is a defining principle of scientific research, and broadly refers to the ability of researchers, other than the original researchers, to achieve the same findings using the same data and analysis data reproducibility is key to the reinforcement and credibility of scientific evidence. All results should be replicable by different investigators in varied geographical settings, using independent data, instruments, and analytical methods.
Some examples:
In 2022 there were 11 Federal Register notices with ORI findings of research misconduct that involved Public Health Service support or funding. These cases included falsified data submitted in National Institutes of Health grant applications and PHS-supported publications. These cases resulted debarment periods of up to four years and supervision periods of up to 12 years.
Without a doubt it is critical to build a quality culturewithin our research organizations. Through educating our scientific staff we can continue to innovate and discover new pathways, new drugs and new treatments. Efficient processes enhance research effectiveness and lead to scientific discoveries. Data integrity supports good science, drug safety, products and treatment development for patients and customers. While this looks similar in research as in later phases there are 4 primary pillars:
Train researchers on basic documentation processes and good scientific practices to ensure data integrity and quality. Targeted training should be added on new guidelines, processes and regulations applied to their specific activities.
Bespalov, A., Bernard, R., Gilis, A., Gerlach, B., Guillén, J., Castagné, V., Lefevre, I. A., Ducrey, F., Monk, L., Bongiovanni, S., Altevogt, B., Arroyo-Araujo, M., Bikovski, L., Bruin, N. de, Castaños-Vélez, E., Dityatev, A., Emmerich, C. H., Fares, R., Ferland-Beckham, C., … Steckler, T. (2021, May 24). Introduction to the EQIPD Quality System. eLife. https://elifesciences.org/articles/63294
There is no term more misused and misunderstood than “Phase Appropriate.” It is one of those terms that just about everyone involved in FDA-regulated industries has an opinion on and one where we all get tripped up.
What do we mean by phase?
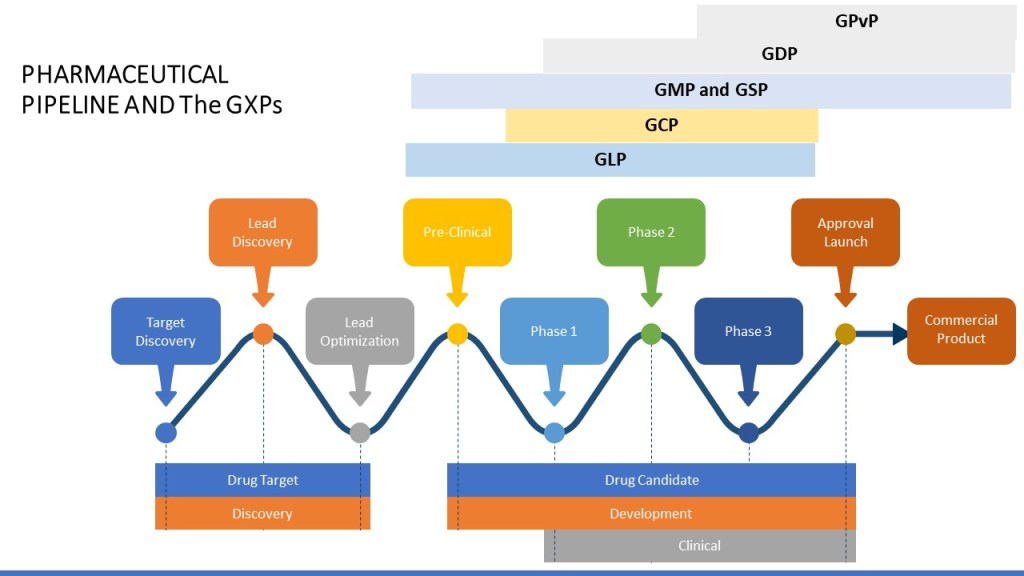
Drug development can be divided into discovery, preclinical studies, clinical development, and market approval.
Each one of these phases is further broken down.
It is also important to remember that certain activities may start in earlier phases. For example, for manufacturing, tech transfer, and commercial manufacturing can start in Phase 3 (and more and more these days even 2!).
An investigational drug for use in a phase 1 study, as described in § 312.21(a) of this chapter, is subject to the statutory requirements set forth in 21 U.S.C. 351(a)(2)(B). The production of such drug is exempt from compliance with the regulations in part 211 of this chapter. However, this exemption does not apply to an investigational drug for use in a phase 1 study once the investigational drug has been made available for use by or for the sponsor in a phase 2 or phase 3 study, as described in § 312.21(b) and (c) of this chapter, or the drug has been lawfully marketed. If the investigational drug has been made available in a phase 2 or phase 3 study or the drug has been lawfully marketed, the drug for use in the phase 1 study must comply with part 211.
Guideline on the responsibilities of the sponsor with regard to handling and shipping of investigational medicinal products for human use in accordance with Good Clinical Practice and Good Manufacturing Practice
Processes characterized and Production and Process Controls (PPC) identified
Analytical methods are qualified
Materials acceptance criteria
Critical vendors qualified
Phase 3:
Processes validated with Production and Process Controls (PPC) identified and controlled
Validation of analytical methods
Materials have been fully qualified and tested upon receipt as appropriate
What About the Quality System?
ICH Q10 clearly spells out the PQS requirements, breaking down into stages of Pharmaceutical Development (usually Phase 1 and earlier), Technology Transfer (usually phase 2), Commercial Manufacturing (which may start before approval) and Product Discontinuation. Q10 then lays out the expectations by these stages for the four key elements of:
Process performance and product quality monitoring system
Corrective action and preventive action (CAPA) system
Change management system
Management review of process performance and product quality.
Pharmaceutical Development
Technology Transfer
Commercial Manufacturing
Product Discontinuation
Process Performance and Product Quality
Process and product knowledge generated and process and product monitoring conducted throughout development can be used to establish a control strategy for manufacturing.
Monitoring during scale-up activities can provide a preliminary indication of process performance and the successful integration into manufacturing. Knowledge obtained during transfer and scale up activities can be useful in further developing the control strategy.
A well-defined system for process performance and product quality monitoring should be applied to assure performance within a state of control and to identify improvement areas.
Once manufacturing ceases, monitoring such as stability testing should continue to completion of the studies. Appropriate action on marketed product should continue to be executed according to regional regulations.
Corrective Action and Preventive Action
Product or process variability is explored. CAPA methodology is useful where corrective actions and preventive actions are incorporated into the iterative design and development process.
CAPA can be used as an effective system for feedback, feedforward and continual improvement.
CAPA should be used and the effectiveness of the actions should be evaluated.
CAPA should continue after the product is discontinued. The impact on product remaining on the market should be considered as well as other products which might be impacted.
Change Management
Change is an inherent part of the development process and should be documented; the formality of the change management process should be consistent with the stage of pharmaceutical development.
The change management system should provide management and documentation of adjustments made to the process during technology transfer activities.
A formal change management system should be in place for commercial manufacturing. Oversight by the quality unit should provide assurance of appropriate science and risk based assessments.
Any changes after product discontinuation should go through an appropriate change management system.
Management Review of Process Performance and Product Quality
Aspects of management review can be performed to ensure adequacy of the product and process design.
Aspects of management review should be performed to ensure the developed product and process can be manufactured at commercial scale.
Management review should be a structured system, as described above, and should support continual improvement.
Management review should include such items as product stability and product quality complaints.
ICH Stage appropriate quality system elements
Together with ICH Q9, this sets forth a framework of building knowledge and risk management into all aspects of the system together with a robust issue management mindset. There are really three things driving this.
Consistency in execution
Document decision making
Follow through
Some aspects remain pretty steady in all phases/stages, while others will grow as the organization develops.
The Difference Between Maturity and Phase Appropriate
People confuse phase appropriate with maturity all the time. Phase appropriate means doing the right activities in the right order. Maturity means the how is the most effective possible.
Quality Management Maturity (QMM) is the state attained when drug manufacturers have consistent, reliable, and robust business processes to achieve quality objectives and promote continual improvement. This is both composed of phase independent and phase dependent aspects.
Remember, a Quality Culture is the foundation that makes the rest of this happen.
Jargon is something we should work hard to avoid, and yet there is an awful lot of it we find difficult to let go. Right at the top is the GxPs.
GxP is a general abbreviation for the “good practice” quality guidelines and regulations. The “x” stands for the various fields, including the pharmaceutical and food industries, for example good manufacutiring practice, or GMP.
There are a lot of GxPs, though we tend to focus on 5(ish), depending on where you are.
We tend to argue a lot about them. Even to the GxP vs GXP. Or GPvP vs GVP. Or GdocP or GDP (so damn confusing, there is another GDP – Good Distribution Practices). Or if Good Storage Practice is its own body or part of the GMPs and GDPs. And…and…and.. The arguing can be fun.
The Five big ones in pharma and medical devices are GLP, GCP, GMP, GDP and GPvP. Some of the others like GACP are pretty intesting in their application.
Some like GDocP and GAMP are more specific threads that go across the GxPs.
By nature the GxPs are tied to the phase of the pharmaceutical pipeline.
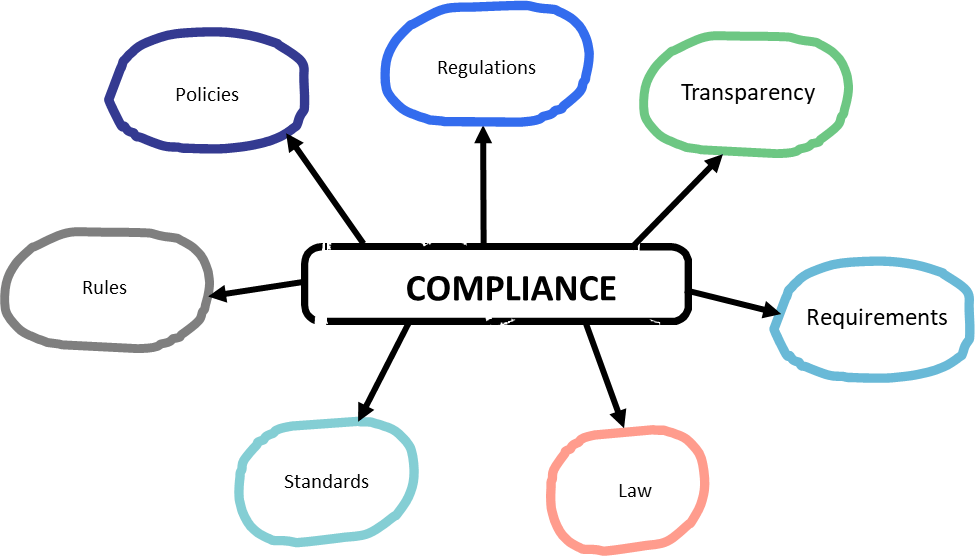
The GxPs are all about ensuring compliance and are informed from a wide range of sources, starting with law and regulations.
Being in the age of globalization, there are many many sources to draw from.
This can also draw from beyond the health authorities (for example in the US USDA for GACP or the DEA for parts of the GDPs).
At the end of the day, GxPs answer to five important criteria.
The quality profession in the pharmaceutical industry is wider than the overseeing regulations from health authorities. The GXPs are truly a starting point, not an ending point which is why we often use that little “c” for “current.”
The GXPs (and the regulations behind them) serve an important purpose. But they are the start of excellence and not the end.
The GXPs are an outline. When we start with these requirements and then fill in the details we build a robust and beautiful engine for quality.
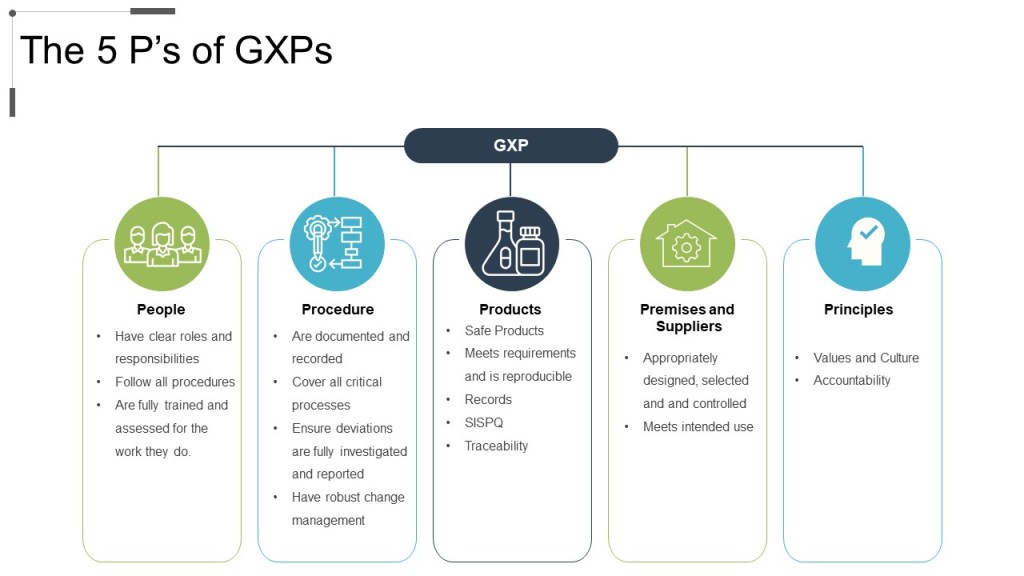
The 5Ps of GXPs: People, Procedure, Product, Premise, and Principles.
This is often why we talk about compliance being the start of quality, and not the end.