In recent years, the importance of diversity in clinical trials has gained significant attention in the medical research community. This focus is not just a matter of inclusivity; it’s a crucial scientific and ethical imperative that directly impacts the quality and applicability of medical research.
Why Diversity in Clinical Trials is Essential
Scientific Validity and Generalizability
Different populations may respond differently to the same treatment due to variations in genetics, lifestyle, and environmental factors. By including diverse participants, researchers can better understand how a treatment works across various groups, leading to more accurate and widely applicable results.
Addressing Health Disparities
Minority groups often experience poorer health outcomes in various diseases. Including these groups in clinical trials is a crucial step towards understanding and addressing these disparities, potentially leading to more targeted and effective treatments for underserved populations.
Innovation and Discovery
Diversity in clinical trials can lead to unexpected discoveries. For instance, the identification of PCSK9, which revolutionized our understanding of cholesterol homeostasis, was a result of studying variations in cardiovascular risk factors among different racial groups.
Alignment with ICH Guidelines
The International Council for Harmonisation (ICH) has recognized the importance of diversity in its updated guidelines, particularly in ICH E6(R3) and ICH E8(R1).
ICH E6(R3)
This guideline emphasizes the importance of including diverse patient populations in clinical trials. It encourages the use of innovative trial designs and technologies to enable wider participation and inclusion of diverse populations. The guideline also stresses the need for quality by design (QbD) and a focus on critical-to-quality factors, which inherently includes considerations of diversity to ensure the reliability of trial results.
ICH E8(R1)
ICH E8(R1) focuses on the general considerations for clinical studies and emphasizes the importance of engaging with a broader range of stakeholders, including patients and patient advocacy groups. This approach naturally leads to more diverse perspectives in trial design and conduct, potentially increasing participation from underrepresented groups.
The Impact of Recent Policy Changes
The recent purge of FDA pages on clinical trial diversity, as reported by STAT News, raises significant concerns about the future of inclusive clinical. This action, part of a wider executive order banning diversity, equity, and inclusion (DEI) initiatives, could have far-reaching consequences:
Reduced Guidance: The removal of these resources may leave researchers and pharmaceutical companies with less clear direction on how to ensure diverse representation in their trials.
Potential Setbacks: Years of progress in improving trial diversity could be undermined, potentially leading to less representative studies and, consequently, less generalizable results.
Health Equity Concerns: This move could exacerbate existing health disparities by reducing the focus on including underrepresented groups in clinical research.
Scientific Integrity: The quality and applicability of clinical trial data may be compromised if diversity is not actively pursued, potentially affecting the safety and efficacy of new treatments for certain populations.
Moving Forward
Despite this setback, the scientific and pharma community must continue to prioritize diversity in clinical trials. The principles outlined in ICH E6(R3) and E8(R1) provide a strong foundation for this effort. Researchers, pharmaceutical companies, and regulatory bodies should:
Continue to develop innovative recruitment strategies to reach diverse populations.
Engage with community leaders and organizations to build trust and awareness about clinical trials.
Design trials with flexibility to improve access for all populations, including the use of decentralized trial elements.
Maintain a focus on quality by design, ensuring that diversity considerations are built into trial planning from the outset.
It is important to remember that E6(r3) is the regulation in Europe, while it is a guidance in the US. So companies need to follow it for their EMA approval possibilities.
In conclusion, diversity in clinical trials is not just a matter of equity; it’s a scientific necessity that ensures the development of safe and effective treatments for all populations. While recent policy changes may present challenges, the medical research community must remain committed to this crucial aspect of clinical research, guided by international standards and ethical imperatives.
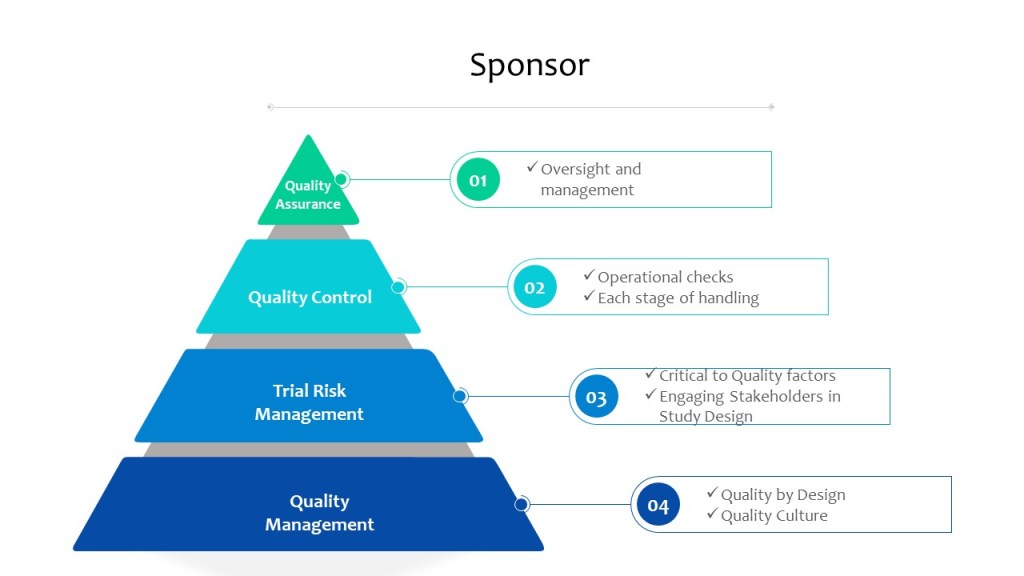
With ICH E6(r3) in draft, I think it is important to look at the current state of risk management expectations in a clinical study.
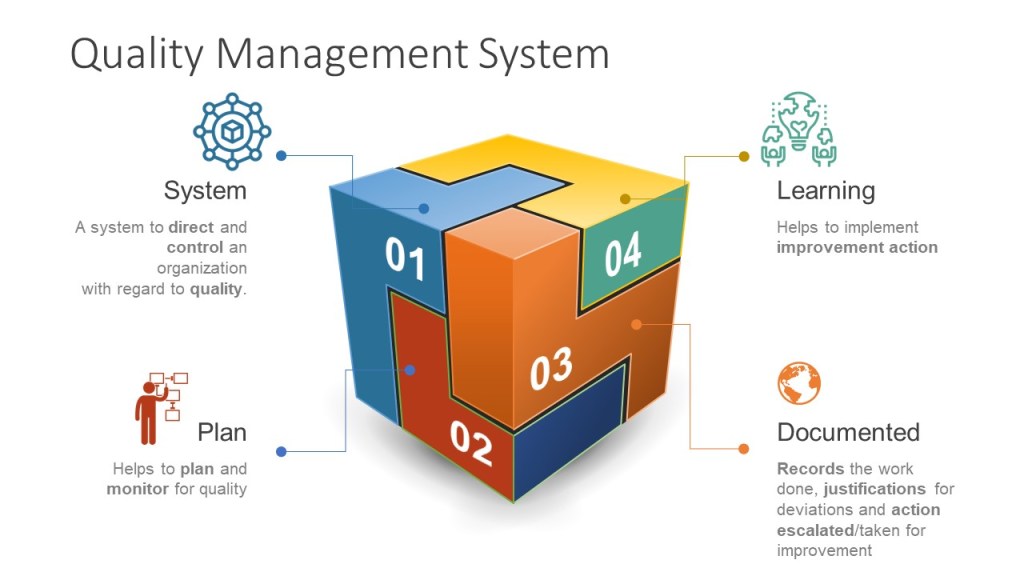
Risk management is an essential part of any clinical study, and is a critical component of the ICH E6 and E8 guidelines for Good Clinical Practice (GCP). These guidelines provide a framework for ensuring the safety and well-being of study participants, as well as the integrity and reliability of the study data. By following the principles outlined in these guidelines, researchers can help to ensure that their study results are reliable and can be used to inform clinical practice.
Through risk management we ensure the four main goals of the GCPs are obtained.
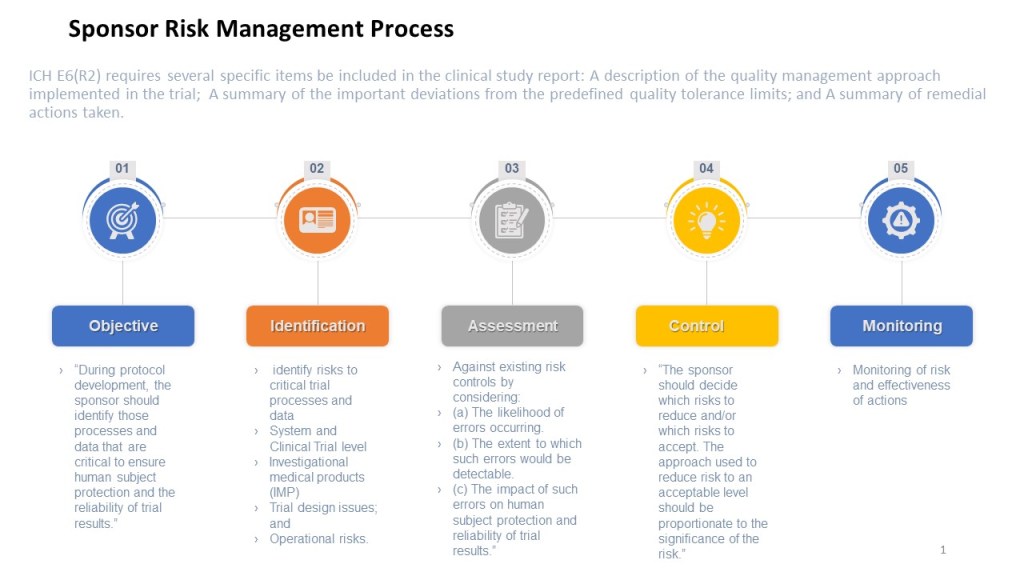
The ICH E6 guideline provides recommendations for the conduct of clinical trials, emphasizing the importance of risk management, specifying that a risk management plan should be developed and implemented for each study. The guideline also provides recommendations for the content of the risk management plan, including the identification of potential risks, the assessment of their likelihood and potential impact, and the development of strategies for managing or mitigating those risks.
Risk management is a key enabler and result of the quality management system.
The ICH E8 guideline, which focuses on the conduct of clinical trials also emphasizes the importance of risk management. It specifies that the risk management plan should include a comprehensive evaluation of the risks associated with the study interventions, as well as a plan for managing or mitigating those risks. The guideline also recommends that the risk management plan be regularly reviewed and updated as needed, to ensure that it continues to effectively address the risks facing the study.
When planning a clinical study, sponsors must carefully consider the potential risks involved and take steps to minimize them. Sources of the risk assessment include performing a thorough literature review to identify any known risks associated with the study interventions, as well as conducting pre-study assessments to identify potential risks specific to the study population. E8 also state sthe importance of a wide variety of stakeholders, including the patient population.
Once the study is underway, it’s important to closely monitor for potential risks and have a plan in place for managing them.
In addition to protecting the safety of study participants, effective risk management is also essential for maintaining the integrity of the data being collected. Risks to the study data might include things like errors in data entry or missing data, which can compromise the validity of the study results. To address these risks, sponsors must have robust quality control measures in place, such as regular data audits and checks for missing or inconsistent data.
Overall, the role of risk management in a clinical study is to ensure the safety and well-being of study participants, while also protecting the integrity of the data being collected. By carefully considering and managing potential risks, researchers can help to ensure that their study results are reliable and can be used to inform clinical practice.
Risk Based Monitoring
Risk-based monitoring is a approach to monitoring the quality of a clinical study that focuses on identifying and addressing potential risks to the study. This approach involves regularly assessing the risks associated with a study and implementing strategies to manage or mitigate those risks.
In a risk-based monitoring approach, the study team typically uses a risk register to identify and assess potential risks to the study, such as the potential for errors in data collection or analysis, or the potential for adverse events in study participants. The team then develops a plan for addressing these risks, which might involve implementing additional quality control measures or training for study staff.
During the study, the team regularly monitors for potential risks and takes action to address them as needed. This might involve conducting regular audits or reviews of the study data to identify potential errors, or monitoring the health and well-being of study participants to identify and address any adverse events.
Overall, the goal of risk-based monitoring is to ensure the quality and integrity of a clinical study by proactively identifying and addressing potential risks. By using a risk-based approach, the study team can help to ensure that the study results are reliable and can be used to inform clinical practice.
Risk Register
A risk register is a document that is used to identify, assess, and track potential risks in a clinical study. It typically includes a list of identified risks, along with information about their likelihood and potential impact, as well as the actions that are being taken to manage or mitigate the risks.
In a clinical study, a risk register might include risks such as the potential for errors in data collection or analysis, the potential for adverse events in study participants, or the potential for the study to be impacted by external factors, such as changes in regulatory requirements.
The purpose of a risk register in a clinical study is to help the study team identify and prioritize potential risks, and to develop strategies for addressing them. By having a clear and comprehensive overview of the risks that a study is facing, the team can take proactive steps to manage or mitigate those risks, and can monitor their progress over time.
Overall, a risk register is an essential tool for managing risks in a clinical study. By providing a clear and comprehensive overview of potential risks, it helps the study team identify and address risks in a proactive and effective way.
Identifying potential risks: The first step in implementing a clinical risk management program is to identify potential risks to the study, such as the potential for errors in data collection or analysis, or the potential for adverse events in study participants. This might involve reviewing the study protocol and data collection tools, consulting with the study team and other stakeholders, and conducting a thorough assessment of the study environment.
Assessing risks: Once potential risks have been identified, the next step is to assess their likelihood and potential impact. This will help to prioritize the risks and determine the appropriate level of response. For example, a risk with a high likelihood and a high potential impact might require more immediate action, while a risk with a low likelihood and a low potential impact might not require as much attention.
Developing strategies for managing risks: Based on the assessment of risks, the next step is to develop strategies for managing or mitigating those risks. This might involve implementing additional quality control measures, providing training to study staff, or conducting regular audits or reviews of the study data. The goal is to develop a comprehensive and effective plan for addressing the identified risks.
Monitoring for potential risks: Once the risk management plan is in place, it’s important to regularly monitor for potential risks and take action to address them as needed. This might involve conducting regular audits or reviews of the study data, or monitoring the health and well-being of study participants. By proactively monitoring for potential risks, the study team can help to ensure the safety and well-being of study participants, as well as the integrity and reliability of the study data.
Follow-up and corrective action: If potential risks are identified during the study, it’s important to take prompt action to address them. This might involve implementing corrective action plans, such as retraining study staff or revising the study protocol. It’s also important to track the progress of these plans and ensure that they are effective in addressing the identified risks. By taking timely and effective action to address potential risks, the study team can help to ensure the safety and well-being of study participants, as well as the integrity and reliability of the study data.
Risk Management in the Clinical Study Process
To summarize, each clinical study should:
Identify Risks
Before the study begins, the sponsor should perform a thorough review of the study protocol, data collection tools, and other study-related documents to identify potential risks to the study.
The cross-functional study team, CROs and other relevant stakeholders, such as the sponsor and regulatory authorities, to identify additional potential risks.
All identified risks should be documented in the study’s risk register.
2. Assess Risks
For each identified risk, assess its likelihood and potential impact on the study.
The risks should be prioritized based on their likelihood and potential impact, with a focus on the highest-priority risks.
3. Manage Risks
For each identified risk, the sponsor should develop a plan for managing or mitigating the risk. This plan should be documented in the study’s risk register.
The plan for managing or mitigating each risk should include specific actions to be taken, as well as the individuals or groups responsible for implementing those actions.
4. Monitor Risks
Regularly monitor key risk indicators and the study for success of the study risk plan and to identify new potential risks and take action to address them as needed. This might involve conducting regular audits or reviews of the study data, or monitoring the health and well-being of study participants.
Any significant risks that arise during the study should be reported to the sponsor and relevant regulatory authorities.
Let us turn our failure space model, and level of problems, to deviations in a clinical trial. This is one of those areas that regulations and tribal practice have complicated, perhaps needlessly. It is also complicated by the different players of clinical sites, sponsor, and usually these days a number of Contract Research Organizations (CRO).
What is a Protocol Deviation?
Protocol deviation is any change, divergence, or departure from the study design or procedures defined in the approved protocol.
Protocol deviations may include unplanned instances of protocol noncompliance. For example, situations in which the clinical investigator failed to perform tests or examinations as required by the protocol or failures on the part of subjects to complete scheduled visits as required by the protocol, would be considered protocol deviations.
In the case of deviations which are planned exceptions to the protocol such deviations should be reviewed and approved by the IRB, the sponsor, and by the FDA for medical devices, prior to implementation, unless the change is necessary to eliminate apparent immediate hazards to the human subjects (21 CFR 312.66), or to protect the life or physical well-being of the subject (21 CFR 812.150(a)(4)).
The FDA, July 2020. Compliance Program Guidance Manual for Clinical Investigator Inspections (7348.811).
In assessing protocol deviations/violations, the FDA instructs field staff to determine whether changes to the protocol were: (1) documented by an amendment, dated, and maintained with the protocol; (2) reported to the sponsor (when initiated by the clinical investigator); and (3) approved by the IRB and FDA (if applicable) before implementation (except when necessary to eliminate apparent immediate hazard(s) to human subjects).
Regulation/Guidance
States
ICH E-6 (R2) Section 4.5.1-4.5.4
4.5.1“trial should be conducted in compliance with the protocol agreed to by the sponsor and, if required by the regulatory authorities…” 4.5.2 The investigator should not implement any deviation from, or changes of, the protocol without agreement by the sponsor and prior review and documented approval/favorable opinion from the IRB/IEC of an amendment, except where necessary to eliminate an immediate hazard(s) to trial subjects, or when the change(s) involves only logistical or administrative aspects of the trial (e.g., change in monitor(s), change of telephone number(s)). 4.5.3 The investigator, or person designated by the investigator, should document and explain any deviation from the approved protocol. 4.5.4 The investigator may implement a deviation from, or a change in, the protocol to eliminate an immediate hazard(s) to trial subjects without prior IRB/IEC approval/favorable opinion.
ICH E3, section 9.6
The sponsor should describe the quality management approach implemented in the trial and summarize important deviations from the predefined quality tolerance limits and remedial actions taken in the clinical study report
21CFR 312.53(vi) (a)
investigators selected “Will conduct the study(ies) in accordance with the relevant, current protocol(s) and will only make changes in a protocol after notifying the sponsor, except when necessary to protect the safety, the rights, or welfare of subjects.”
21CFR 56.108(a)
IRB shall….ensur[e] that changes in approved research….may not be initiated without IRB review and approval except where necessary to eliminate apparent immediate hazards to the human subjects.
21 CFR 56.108(b)
“IRB shall….follow written procedures for ensuring prompt reporting to the IRB, appropriate institutional officials, and the Food and Drug Administration of… any unanticipated problems involving risks to human subjects or others…[or] any instance of serious or continuing noncompliance with these regulations or the requirements or determinations of the IRB.”
45 CFR 46.103(b)(5)
Assurances applicable to federally supported or conducted research shall at a minimum include….written procedures for ensuring prompt reporting to the IRB….[of] any unanticipated problems involving risks to subjects or others or any serious or continuing noncompliance with this policy or the requirements or determinations of the IRB.
FDA Form-1572 (Section 9)
lists the commitments the investigator is undertaking in signing the 1572 wherein the clinical investigator agrees “to conduct the study(ies) in accordance with the relevant, current protocol(s) and will only make changes in a protocol after notifying the sponsor, except when necessary to protect the safety, the rights, or welfare of subjects… [and] not to make any changes in the research without IRB approval, except where necessary to eliminate apparent immediate hazards to the human subjects.”
A few key regulations and guidances (not meant to be a comprehensive list)
How Protocol Deviations are Implemented
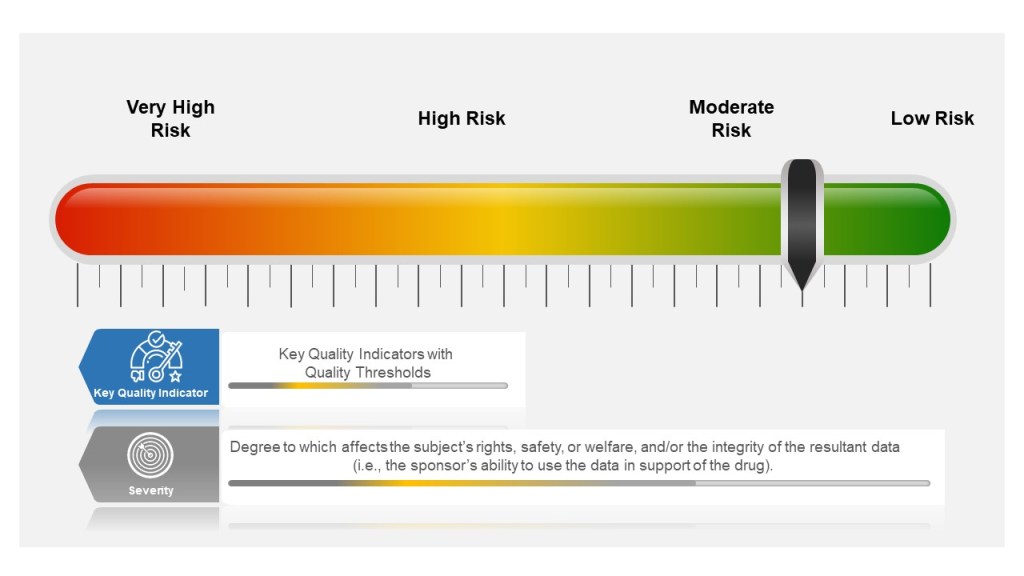
Many companies tend to have a failure scale built into their process, differentiating between protocol deviations and violations based on severity. Others use a minor, major, and even critical scale to denote differences in severity. The axis here for severity is the degree to which affects the subject’s rights, safety, or welfare, and/or the integrity of the resultant data (i.e., the sponsor’s ability to use the data in support of the drug).
Other companies divide into protocol deviations and violations:
Protocol Deviation: A protocol deviation occurs when, without significant consequences, the activities on a study diverge from the IRB-approved protocol, e.g., missing a visit window because the subject is traveling. Not as serious as a protocol violation.
Protocol Violation: A divergence from the protocol that materially (a) reduces the quality or completeness of the data, (b) makes the ICF inaccurate, or (c) impacts a subject’s safety, rights or welfare. Examples of protocol violations may include: inadequate or delinquent informed consent; inclusion/exclusion criteria not met; unreported SAEs; improper breaking of the blind; use of prohibited medication; incorrect or missing tests; mishandled samples; multiple visits missed or outside permissible windows; materially inadequate record-keeping; intentional deviation from protocol, GCP or regulations by study personnel; and subject repeated noncompliance with study requirements.
This is probably a place when nomenclature can serve to get in the way, rather than provide benefit. The EMA says pretty much the same in “ICH guideline E3 – questions and answers (R1).“
Principles of Events in Clinical Practice
Severity of the event is based on degree to which affects the subject’s rights, safety, or welfare, and/or the integrity of the resultant data
Events happen beyond the Protocol. These need to be managed appropriately as well.
The event needs to be categorized, evaluated and trended by the sponsor
Severity of the Event
Starting in the study planning stage, ICH E6(R2) GCP requires sponsors to identify risks to critical study processes and study data and to evaluate these risks based on likelihood, detectability and impact on subject safety and data integrity.
Sponsors then establish key quality indicators (KQIs) and quality tolerance thresholds. KQI is really just a key risk indicator and should be treated similarly.
Study events that exceed the risk threshold should trigger an evaluation to determine if action is needed. In this way, sponsors can proactively manage risk and address protocol noncompliance.
The best practice here is to have a living risk assessment for each study. Evaluate across studies to understand your overall organization risk, and look for opportunities for wide-scale mitigations. Feedup into your risk register.
Event Classification for Clinical Protocols and GCPs
Where the Event happens
Deviations in the clinical space are a great example of the management of supplier events, and at the end of the day there is little difference between a GMP supplier event management, a GLP or a GCP. The individual requirements might be different but the principles and the process are the same.
Each entity in the trial organization should have their own deviation system where they investigate deviations, performing root cause investigation and enacting CAPAs.
This is where it starts to get tricky. first of all, not all sites have the infrastructure to do this well. Second the nature of reporting, usually through the Electronic Data Capture (EDC) system, can lead to balkanization at the site. Site’s need to have strong compliance programs through compiling deviation details into a single sitewide system that allows the site to trend deviations across studies in addition to following sponsor reporting requirements.
Unfortunately too many site’s rely on the sponsor’s program. Sponsors need to be evaluating the strength of this program during site selection and through auditing.
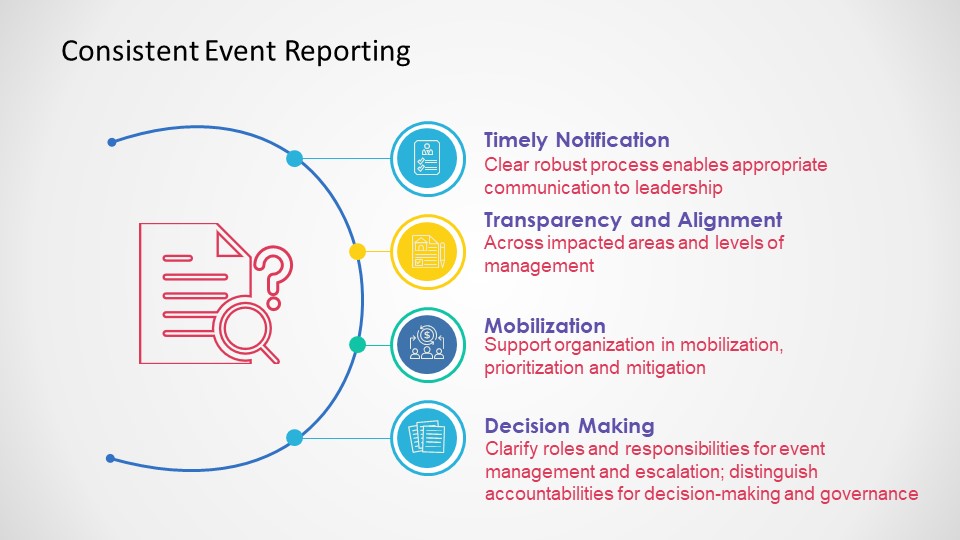
Events Happen
Consistent Event Reporting is Critical
Deviations should be to all process, procedure and plans, and just not the protocol.
Categorization and Trending
Categorizing deviations is usually a pain point and an area where more consistency needs to be driven. I recommend first having a good standard set of categorizations. The industry would benefit from adopting a standard, and I think Norman Goldfarb’s proposal is still the best.
Once you have categories, and understand to your KQIs and other aspects you need to make sure they are consistently done. The key mechanisms of this are: