One of the many fascinating items in the recent Warning Letter to Sanofi is the FDA’s direction to provide a plan to perform “timely technological upgrades to the equipment/facility infrastructure.” This point drives home the point that staying current with technological advancements is crucial for maintaining compliance, improving efficiency, and ensuring product quality. Yet, I think it is fair to say we rarely see it this bluntly put as a requirement.
One of the many reasons this Warning Letter stands out is that this is (as far as I can tell) the same facility that won the ISPE’s Facility of the Year award in 2020. This means it is still a pretty new facility, and since it is one of the templates that many single-use biotech manufacturing facilities are based on, we had best pay attention. If a failure to maintain a state-of-the-art facility can contribute to this sort of Warning Letter, then many companies had best be paying close attention. There is a lot to unpack and learn here.
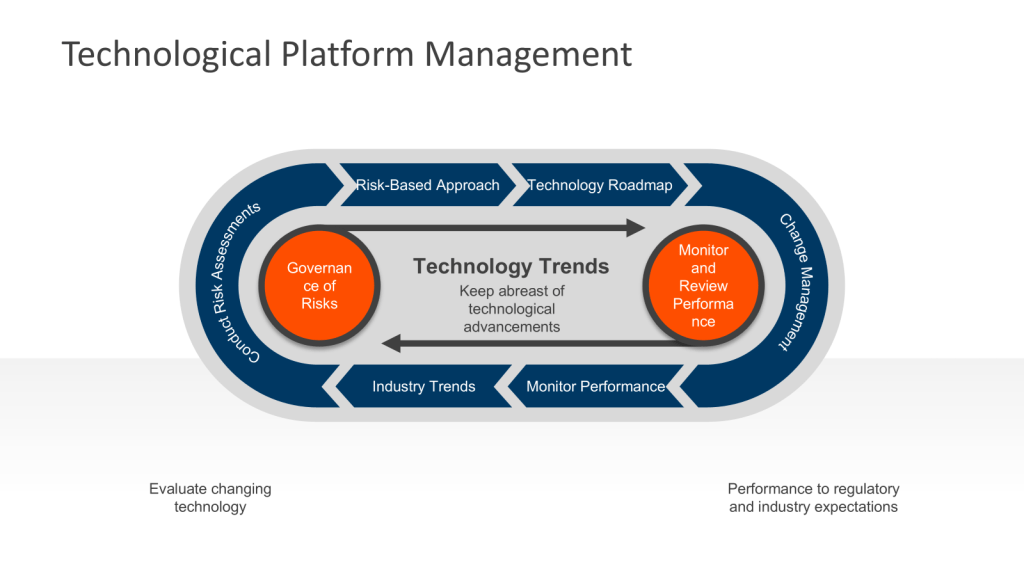
Establishing an Ongoing Technology Platform Process
To meet regulatory requirements and industry standards, facilities should implement a systematic approach to technological upgrades.
1. Conduct Regular Assessments
At least annually, perform comprehensive evaluations of your facility’s equipment, systems, and processes. This assessment should include:
Prioritize upgrades based on their potential impact on product quality, patient safety, and regulatory compliance. Utilize living risk assessments to get a sense of where issues are developing. These should be the evolution of the risk management that built the facility.
4. Create a Technology Roadmap
Develop a long-term plan for implementing upgrades, considering:
Budget constraints and return on investment
Regulatory timelines for submissions and approvals
Production schedules and potential downtime
Integration with existing systems and processes
5. Implement Change Management Procedures
Ensure there is a robust change management process in place to ensure that upgrades are implemented safely and effectively. This should include:
Detailed documentation of proposed changes
Impact assessments on product quality and regulatory compliance
6. Appropriate Verification – Commissioning, Qualification and Validation
Conduct thorough verification activities to demonstrate that the upgraded equipment or systems meet predetermined specifications and regulatory requirements.
7. Monitor and Review Performance
Continuously monitor the performance of upgraded systems and equipment to ensure they meet expectations and comply with cGMP requirements. Conduct periodic reviews to identify any necessary adjustments or further improvements. This is all part of Stage 3 of the FDA’s process validation model focusing on ongoing assurance that the process remains in a state of control during routine commercial manufacture. This stage is designed to:
Anticipate and prevent issues before they occur
Detect unplanned deviations from the process
Identify and correct problems
Leveraging Advanced Technologies
To stay ahead of regulatory expectations and industry trends, consider incorporating advanced technologies into your upgrade plans:
Single-Use Systems (SUS): Implement disposable components to reduce cleaning and validation requirements while improving flexibility.
Modern Microbial Methods (MMM): Implement advanced techniques used in microbiology that offer significant advantages over traditional culture-based methods
Process Analytical Technology (PAT): Integrate real-time monitoring and control systems to enhance product quality and process understanding.
Data Analytics and Artificial Intelligence: Implement advanced data analysis tools to identify trends, predict maintenance needs, and optimize processes.
Conclusion
Maintaining a state-of-the-art biotech facility requires a proactive and systematic approach to technological upgrades. By establishing an ongoing process for identifying and implementing improvements, facilities can ensure compliance with FDA requirements, align with industry standards, and stay competitive in the rapidly evolving biotech landscape.
Remember that the goal is not just to meet current regulatory expectations but to anticipate future requirements and position your facility at the forefront of biotech manufacturing excellence. By following this comprehensive approach and staying informed on industry developments, you can create a robust, flexible, and compliant manufacturing environment that supports the production of high-quality biopharmaceutical products.
Track the time taken to approve validation documents
Supplier Performance
Monitor supplier audit results related to validated systems or components
Track supplier-related deviations or non-conformances
Regulatory Inspection Outcomes
Track the number and severity of validation-related observations during inspections
Measure the time taken to address and close out regulatory findings
Cost and Efficiency Metrics
Measure the time and resources required to complete validation activities
Track cost savings achieved through optimized CQV approaches
By tracking these metrics, we might be able to demonstrate a comprehensive and effective CQV program that aligns with regulatory expectations. Or we might just spend time measuring stuff that may not be tailored to our individual company’s processes, products, and risk profile. And quite frankly, will they influence the system the way we want? It’s time to pull out an IMPACT key behavior analysis to help us tailor a right-sized set of metrics.
The first thing to do is to go to first principles, to take a big step back and ask – what do I really want to improve?
The purpose of a CQV program is to provide documented evidence that facilities, systems, equipment and processes have been designed, installed and operate in accordance with predetermined specifications and quality attributes:
To verify that critical aspects of a facility, utility system, equipment or process meet approved design specifications and quality attributes.
To demonstrate that processes, equipment and systems are fit for their intended use and perform as expected to consistently produce a product meeting its quality attributes.
To establish confidence that the manufacturing process is capable of consistently delivering quality product.
To identify and understand sources of variability in the process to better control it.
To detect potential problems early in development and prevent issues during routine production.
The ultimate measure of success is demonstrating and maintaining a validated state that ensures consistent production of safe and effective products meeting all quality requirements.
Focusing on the Impact is important. What are we truly concerned about for our CQV program. Based on that we come up with two main factors:
The level of deviations that stem from root causes associated with our CQV program
The readiness of FUSE elements for use (project adherence)
Reducing Deviations from CQV Activities
First, we gather data, what deviations are we looking for? These are the types of root causes that we will evaluate. Of course, your use of the 7Ms may vary, this list is to start conversation.
Means
Automation or Interface Design Inadequate/Defective
Validated machine or computer system interface or automation failed to meet specification due to inadequate/defective design.
Means
Preventative Maintenance Inadequate
The preventive maintenance performed on the equipment was insufficient or not performed as required.
Means
Preventative Maintenance Not Defined
No preventive maintenance is defined for the equipment used.
Means
Equipment Defective/Damaged/Failure
The equipment used was defective or a specific component failed to operate as intended.
Means
Equipment Incorrect
Equipment required for the task was set up or used incorrectly or the wrong equipment was used for the task.
Means
Equipment Design Inadequate/Defective
The equipment was not designed or qualified to perform the task required or the equipment was defective, which prevented its normal operation.
Media
Facility Design
Improper or inadequate layout or construction of facility, area, or work station.
Methods
Calibration Frequency is Not Sufficient/Deficiency
Calibration interval is too long and/or calibration schedule is lacking.
Methods
Calibration/Validation Problem
An error occurred because of a data collection- related issue regarding calibration or validation.
Methods
System / Process Not Defined
The system/tool or the defined process to perform the task does not exist.
Based on analysis of what is going on we can move into using a why-why technique to look at our layers.
Why 1
Why are deviations stemming from CQV events not at 0% Because unexpected issues or discrepancies arise after the commissioning, qualification, or validation processes
Success factor needed for this step: Effectiveness of the CQV program
Metric for this step: Adherence to CQV requirements
Why 2 (a)
Why are unexpected issues arising after CQV? Because of inadequate planning and resource constraints in the CQV process.
Success Factor needed for this step: Appropriate project and resource planning
Metric for this Step: Resource allocation
Why 3 (a)
Why are we not performing adequate resource planning? Because of the tight project timelines, and the involvement of multiple stakeholders with different areas of expertise
Success Factor needed for this step: Cross-functional governance to implement risk methodologies to focus efforts on critical areas
Metric for this Step: Risk Coverage Ratio measuring the percentage of identified critical risks that have been properly assessed and and mitigated through the cross-functional risk management process. This metric helps evaluate how effectively the governance structure is addressing the most important risks facing the organization.
Why 2 (b)
Why are unexpected issues arising after CQV? Because of poorly executed elements of the CQV process stemming from poorly written procedures and under-qualified staff.
Success Factor needed for this step: Process Improvements and Training Qualification
Metric for this Step: Performance to Maturity Plan
There were somethings I definitely glossed over there, and forgive me for not providing numbers there, but I think you get the gist.
So now I’ve identified the I – How do we improve reliability of our CQV program, measured by reducing deviations. Let’s break out the rest.
Parameters
Executed for CQV
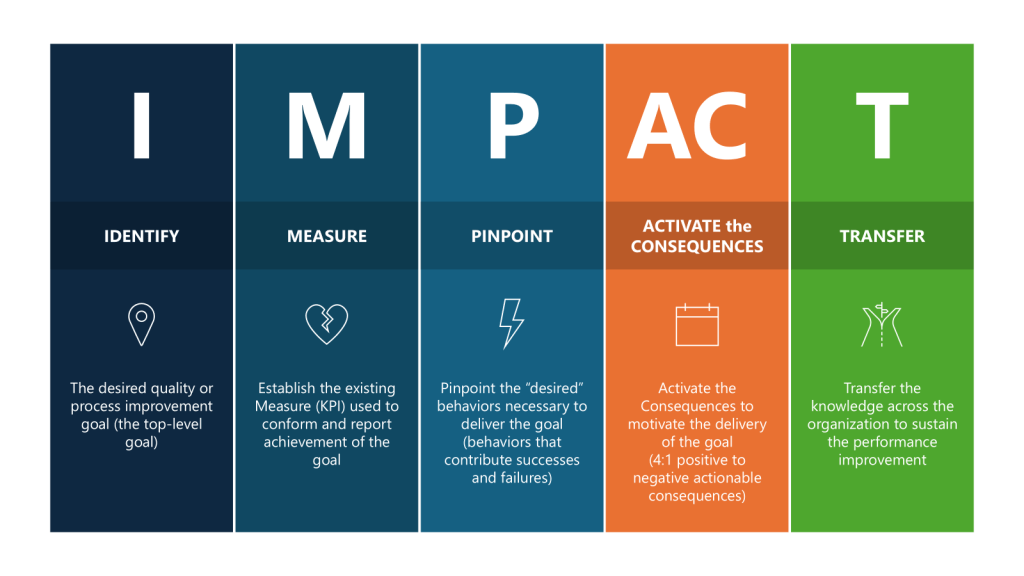
IDENTIFY
The desired quality or process improvement goal (the top-level goal)
Improve the effectiveness of the CQV program by taking actions to reduce deviations stemming from verification of FUSE and process.
MEASURE
Establish the existing Measure (KPI) used to conform and report achievement of the goal
Set a target reduction of deviations related to CQV activities.
Pinpoint
Pinpoint the “desired” behaviors necessary to deliver the goal (behaviors that contribute successes and failures)
Drive good project planning and project adherence.
Promote and coach for enhanced attention to detail where “quality is everyone’s job.”
Encourage a speak-up culture where concerns, issues or suggestions are shared in a timely manner in a neutral constructive forum.
ACTIVATE the CONSEQUENCES
Activate the Consequences to motivate the delivery of the goal (4:1 positive to negative actionable consequences)
Organize team briefings on consequences
Review outcomes of project health
Senior leadership celebrate/acknowledge
Acknowledge and recognize improvements
Motivate the team through team awards
Measure success on individual deliverables through a Rubric
TRANSFER
Transfer the knowledge across the organization to sustain the performance improvement
Create learning teams
Lessons learned are documented and shared
Lunch-and-learn sessions
Create improvement case studies
From these two exercises I’ve now identified my lagging and leading indicators at the KPI and the KBI level.
A key KPI for a FUSE program is Overall Equipment Effectiveness (OEE) which measures the efficiency and productivity of equipment and production processes.
Definition of OEE
OEE is a percentage that represents the proportion of truly productive manufacturing time. It takes into account three main factors:
Availability: The ratio of Run Time to Planned Production Time. It takes into account any events that stop planned production for an appreciable length of time.
Performance: Anything that causes the manufacturing process to run at less than the maximum possible efficiency when it is running.
Quality: Manufactured material that do not meet quality standards, including materialthat require rework and reprocessing.
The formula for calculating OEE is:
OEE = Availability × Performance × Quality
Components of OEE
Availability
Availability measures the percentage of scheduled time that the equipment is available to operate. It accounts for downtime losses.
Availability = Run Time / Planned Production Time
Performance
Performance compares the actual output of equipment to its theoretical maximum output at optimal speed.
Performance = (Ideal Cycle Time × Total Count) / Run Time
Quality
Quality represents the percentage of released material produced out of the total material produced.
Quality = Good Count / Total Count
Importance of OEE
OEE is crucial for several reasons:
It provides a comprehensive view of manufacturing productivity.
It helps identify losses and areas for improvement.
It serves as a benchmark for comparing performance across different equipment or production lines.
It supports continuous improvement initiatives.
Interpreting OEE Scores
While OEE scores can vary by industry, generally:
100% OEE is perfect production
85% is considered world-class
60% is fairly typical
40% is low but not uncommon for companies just starting to measure OEE
Benefits of Tracking OEE
Identifies hidden capacity in manufacturing operations
Reduces manufacturing costs
Improves quality control
Increases equipment longevity through better maintenance practices
Enhances decision-making with data-driven insights
By focusing on OEE, manufacturers can significantly enhance their productivity, reduce waste, and improve their bottom line. It’s a powerful metric that provides actionable insights for optimizing manufacturing processes.
How strongly does this metric connect to business objectives?
5
Empirically Direct – Data proves the metric directly supports at least one business objective – the ability to meet client requirements
Measurability
How much effort would it take to track this metric?
3
Medium – Data exists but in a variety of spreadsheets systems, minor collection or measurement challenges may exist. Will need to agree on what certain aspects of data means.
Precision
How often and by what margin does the metric change?
5
Once we agree on the metric and how to measure it, it should be Highly Predictable
Actionability
Can we clearly articulate actions we would take in response to this metric?
4
Some consensus on action, and capability currently exists to take action. This metric will be used to drive consensus.
Presence of Baseline
Does internal or external baseline data exist to indicate good/poor performance for this metric?
3
Baseline must be based on incomplete or directional data. Quite frankly, the site is just qualified and there will be a rough patch.
This tells me this is a strong metric that requires a fair amount of work to implement. It is certainly going into the Metrics Plan.
A Deeper Dive into Equipment Availability
Equipment availability metric measures the proportion of time a piece of equipment or machinery is operational and ready for production compared to the total planned production time. It is a key component of Overall Equipment Effectiveness (OEE), along with Performance and Quality.
This metric directly impacts production capacity and throughput with a high availability indicating efficient maintenance practices and equipment reliability. This metric helps identify areas for improvement in operations and maintenance.
Definition and Calculation
Equipment availability is expressed as a percentage and calculated using the following formula:
Availability (%) = (Actual Operation Time / Planned Production Time) × 100
Where:
Actual Operation Time = Planned Production Time – Total Downtime
Planned Production Time = Total Time – Planned Downtime
For example, if a machine is scheduled to run for 8 hours but experiences 1 hour of unplanned downtime:
To increase equipment availability, consider the following strategies:
Implement preventive and predictive maintenance programs.
Optimize changeover procedures and reduce setup times.
Enhance operator training to improve equipment handling and minor maintenance skills.
Use real-time monitoring systems to quickly identify and address issues.
Analyze root causes of downtime and implement targeted improvements.
Incorporate fault tolerance at the equipment design stage.
Create asset-specific maintenance programs.
Relationship to Other Metrics
Equipment availability is closely related to other important manufacturing metrics:
It’s one of the three components of OEE, alongside Performance and Quality.
It’s distinct from but related to equipment reliability, which measures the probability of failure-free operation.
It impacts overall plant efficiency and productivity.
By focusing on improving equipment availability, manufacturers can enhance their overall operational efficiency, reduce costs, and increase production capacity. Regular monitoring and analysis of this metric can provide valuable insights for continuous improvement initiatives in manufacturing processes.
To generate an equipment availability KPI in process manufacturing, you should follow these steps:
Calculate Equipment Availability
The basic formula for equipment availability is:
Availability = Run Time / Planned Production Time
Where:
Run Time = Planned Production Time – Downtime
Planned Production Time = Total Time – Planned Downtime
For example, if a machine is scheduled to run for 8 hours, but has 1 hour of unplanned downtime:
To calculate availability accurately, you need to track:
Total available time
Planned downtime (e.g. scheduled maintenance)
Unplanned downtime (e.g. breakdowns)
Actual production time
Implement Data Collection Systems
Use automated data collection systems like machine monitoring software or manufacturing execution systems (MES) to capture accurate, real-time data on equipment status and downtime.
Analyze Root Causes
Categorize and analyze causes of downtime to identify improvement opportunities. Common causes include:
Equipment failures
Changeovers/setups
Material shortages
Operator availability
Set Targets and Monitor Trends
Set realistic availability targets based on industry benchmarks and your current performance
Track availability over time to identify trends and measure improvement efforts
Compare availability across equipment and production lines
Take Action to Improve Availability
Implement preventive and predictive maintenance programs
Optimize changeover procedures
Improve operator training
Address chronic equipment issues
Use Digital Tools
Leverage technologies like IoT sensors, cloud analytics, and digital twins to gain deeper insights into equipment performance and predict potential failures.
Planned Production Time
Planned production time is the total amount of time scheduled for production activities, excluding planned downtime. It represents the time during which equipment or production lines are expected to be operational and producing goods. It can be rather tricky to agree on the exact meaning.
Calculation
The basic formula for planned production time is:
Planned Production Time = Total Time – Planned Downtime
Where:
Total Time is the entire time period being considered (e.g., a shift, day, week, or month)
Planned Downtime includes scheduled maintenance, changeovers, and other planned non-productive activities
Components of Planned Production Time
Total Time
This is the full duration of the period being analyzed, such as:
A single 8-hour shift
A 24-hour day
A 7-day week
A 30-day month
Planned Downtime
This includes all scheduled non-productive time, such as:
Preventive maintenance
Scheduled breaks
Shift changes
Planned changeovers between batches
Cleaning and sanitation procedures
Considerations for Batch Manufacturing
In batch production, several factors affect planned production time:
Batch Changeovers: Time allocated for switching between different product batches must be accounted for as planned downtime.
Equipment Setup: The time required to configure machinery for each new batch should be included in planned downtime.
Quality Checks: Time for quality control procedures between batches may be considered part of planned production time or planned downtime, depending on the specific process.
Cleaning Procedures: Time for cleaning equipment between batches is typically considered planned downtime.
Material Handling: Time for loading raw materials and unloading finished products between batches may be part of planned production time or downtime, based on the specific process.
Example Calculation
Let’s consider a single 8-hour shift in a batch manufacturing facility:
As October rolls around I am focusing on 3 things: finalizing a budget; organization design and talent management; and a 2025 metrics plan. One can expect those three things to be the focus of a lot of my blog posts in October.
Go and read my post on Metrics plans. Like many aspects of a quality management system we don’t spend nearly enough time planning for metrics.
So over the next month I’m going to develop the strategy for a metrics plan to ensure the optimal performance, safety, and compliance of our biotech manufacturing facility, with a focus on:
Facility and utility systems efficiency
Equipment reliability and performance
Effective commissioning, qualification, and validation processes
Robust quality risk management
Stringent contamination control measures
Following the recommended structure of a metrics plan, here is the plan:
Rationale and Desired Outcomes
Implementing this metrics plan will enable us to:
Improve overall facility performance and product quality
Reduce downtime and maintenance costs
Ensure regulatory compliance
Minimize contamination risks
Optimize resource allocation
Metrics Framework
Our metrics framework will be based on the following key areas:
Facility and Utility Systems
Equipment Performance
Commissioning, Qualification, and Validation (CQV)
Quality Risk Management (QRM)
Contamination Control
Success Criteria
Success will be measured by:
Reduction in facility downtime
Improved equipment reliability
Faster CQV processes
Decreased number of quality incidents
Reduced contamination events
Implementation Plan
Steps, Timelines & Milestones
Develop detailed metrics for each key area (Month 1)
Implement data collection systems (Month 2)
Train personnel on metrics collection and analysis (Month 3)
Begin data collection and initial analysis (Month 4)
Review and refine metrics (Month 9)
Full implementation and ongoing analysis (Month 12 onwards)
This plan gets me ready to evaluate these metrics as part of governance in January of next year.
In October I will breakdown some metrics, explaining them and provide the rationale, and demonstrate how to collect. I’ll be striving to break these metrics into key performance indicators (KPI), key behavior indicators (KBI) and key risk indicators (KRI).