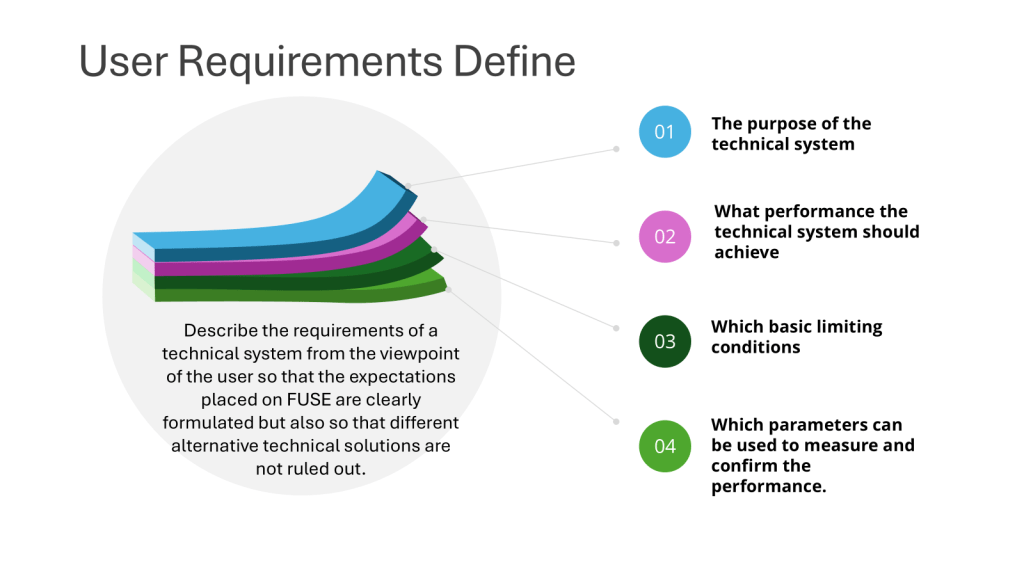
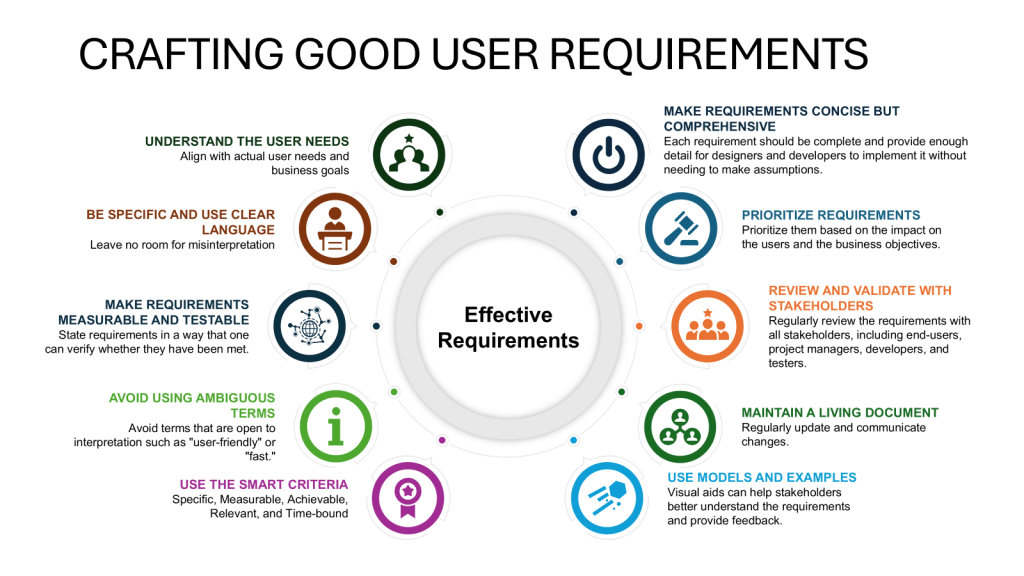
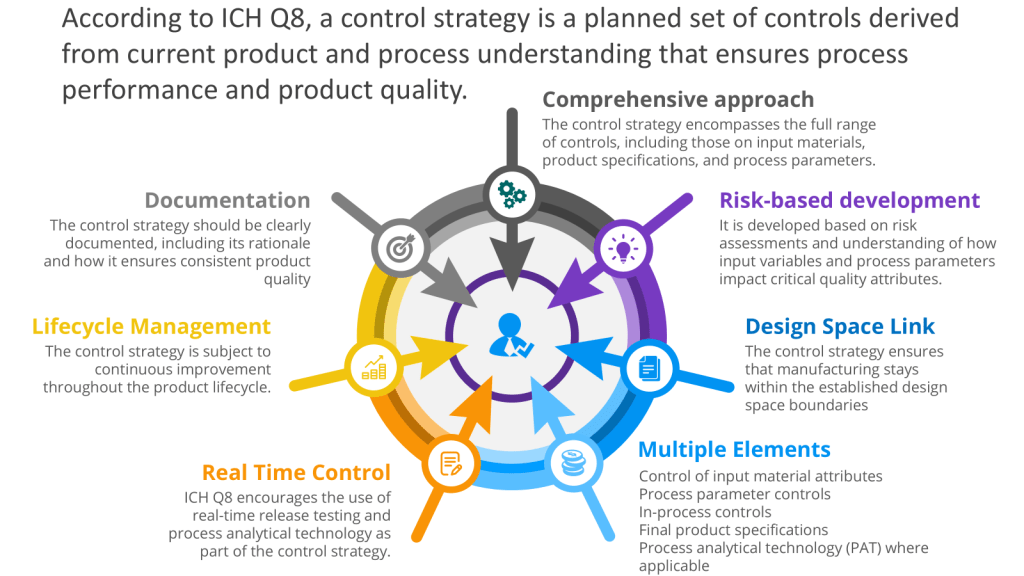
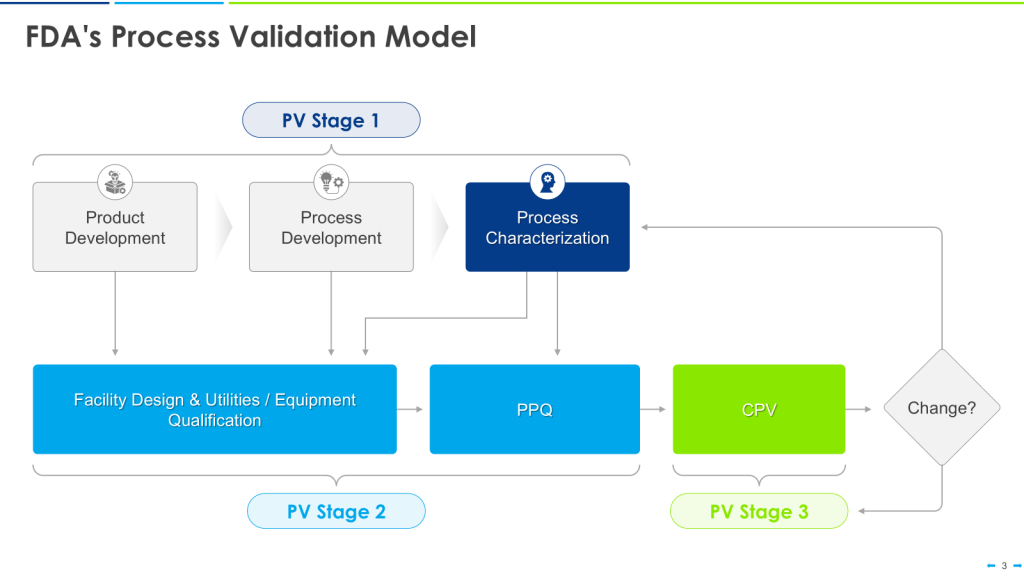
User requirements are typically divided into several categories to ensure comprehensive coverage of all aspects of product development, manufacturing, and quality control and to help guide the risk-based approach to verification.
Quality requirements focus on ensuring that the product meets all necessary quality standards and regulatory compliance. This category includes:
Good Manufacturing Practices (GMP) compliance, including around cleaning, cross-contamination, etc to ensure compliance with various regulations such as FDA guidelines, EU GMP, and ICH standards.
Documentation and record-keeping standards
Contamination control strategies are a key part of quality requirements, as they are essential for maintaining product quality and patient safety.
Data integrity requirements fall under this category, as they are crucial for ensuring the quality and reliability of data.
Not everyone advocates for this breakdown but I am a huge proponent as it divides the product specific requirements for the more standard must’s of meeting the cGMPs that are not product specific. This really helps when you are a multi-product facility and it helps define what is in the PQ versus what is in the PPQ.
Safety User Requirements
Safety requirements address the safety of personnel, patients, and the environment. They encompass:
Occupational health and safety measures
Environmental protection protocols
Patient safety considerations in product design
General User Requirements
General requirements cover broader aspects of the manufacturing system and facility. These may include:
Facility design and layout
Equipment specifications
Utility requirements (e.g., power, water, HVAC)
Maintenance procedures
By categorizing user requirements in this way, pharmaceutical companies can ensure a comprehensive approach to product development and manufacturing, addressing all critical aspects from product quality to regulatory compliance and safety. This will help drive appropriate verification.
“The specification for equipment, facilities, utilities or systems should be defined in a URS and/or a functional specification. The essential elements of quality need to be built in at this stage and any GMP risks mitigated to an acceptable level. The URS should be a point of reference throughout the validation life cycle.” – Annex 15, Section 3.2, Eudralex Volume 4
User Requirement Specifications serve as a cornerstone of quality in pharmaceutical manufacturing. They are not merely bureaucratic documents but vital tools that ensure the safety, efficacy, and quality of pharmaceutical products.
Defining the Essentials
A well-crafted URS outlines the critical requirements for facilities, equipment, utilities, systems and processes in a regulated environment. It captures the fundamental aspects and scope of users’ needs, ensuring that all stakeholders have a clear understanding of what is expected from the final product or system.
The phrase “essential elements of quality need to be built in at this stage” emphasizes the proactive approach to quality assurance. By incorporating quality considerations from the outset, manufacturers can:
Reduce the need for costly corrections later in the process
Ensure compliance with Good Manufacturing Practice (GMP) standards
Mitigating GMP Risks
Risk management is a crucial aspect of pharmaceutical manufacturing. The URS plays a vital role in identifying and addressing potential GMP risks early in the development process. By doing so, manufacturers can:
Ensure that the final product meets regulatory requirements
The URS as a Living Document
One of the key points in the regulations is that the URS should be “a point of reference throughout the validation life cycle.” This underscores the dynamic nature of the URS and its ongoing importance.
Continuous Reference
Throughout the development, implementation, and operation of a system or equipment, the URS serves as:
A benchmark for assessing progress
A guide for making decisions
A tool for resolving disputes or clarifying requirements
Adapting to Change
As projects evolve, the URS may need to be updated to reflect new insights, technological advancements, or changing regulatory requirements. This flexibility ensures that the final product remains aligned with user needs and regulatory expectations.
Practical Implications
Involve multidisciplinary teams in creating the URS, including representatives from quality assurance, engineering, production, and regulatory affairs.
Conduct thorough risk assessments to identify potential GMP risks and incorporate mitigation strategies into the URS.
Ensure clear, objectively stated requirements that are verifiable during testing and commissioning.
Align the URS with company objectives and strategies to ensure long-term relevance and support.
Implement robust version control and change management processes for the URS throughout the validation lifecycle.
Executing the Control Space from the Design Space
The User Requirements Specification (URS) is a mechanism for executing the control space, from the design space as outlined in ICH Q8. To understand that, let’s discuss the path from a Quality Target Product Profile (QTPP) to Critical Quality Attributes (CQAs) to Critical Process Parameters (CPPs) with Proven Acceptable Ranges (PARs), which is a crucial journey in pharmaceutical development using Quality by Design (QbD) principles. This systematic approach ensures that the final product meets the desired quality standards and user needs.
It is important to remember that this is usually a set of user requirements specifications, respecting the system boundaries.
From QTPP to CQAs
The journey begins with defining the Quality Target Product Profile (QTPP). The QTPP is a comprehensive summary of the quality characteristics that a drug product should possess to ensure its safety, efficacy, and overall quality. It serves as the foundation for product development and includes considerations such as:
Dosage strength
Delivery system
Dosage form
Container system
Purity
Stability
Sterility
Once the QTPP is established, the next step is to identify the Critical Quality Attributes (CQAs). CQAs are physical, chemical, biological, or microbiological properties that should be within appropriate limits to ensure the desired product quality. These attributes are derived from the QTPP and are critical to the safety and efficacy of the product.
From CQAs to CPPs
With the CQAs identified, the focus shifts to determining the Critical Process Parameters (CPPs). CPPs are process variables that have a direct impact on the CQAs. These parameters must be monitored and controlled to ensure that the product consistently meets the desired quality standards. Examples of CPPs include:
Temperature
pH
Cooling rate
Rotation speed
The relationship between CQAs and CPPs is established through risk assessment, experimentation, and data analysis. This step often involves Design of Experiments (DoE) to understand how changes in CPPs affect the CQAs. This is Process Characterization.
Establishing PARs
For each CPP, a Proven Acceptable Range (PAR) is determined. The PAR represents the operating range within which the CPP can vary while still ensuring that the CQAs meet the required specifications. PARs are established through rigorous testing and validation processes, often utilizing statistical tools and models.
Build the Requirements for the CPPs
The CPPs with PARs are process parameters that can affect critical quality attributes of the product and must be controlled within predetermined ranges. These are translated into user requirements. Many will specifically label these as Product User Requirements (PUR) to denote they are linked to the overall product capability. This helps to guide risk assessments and develop an overall verification approach.
Most of Us End Up on the Less than Happy Path
This approach is the happy path that aligns nicely with the FDA’s Process Validation Model.
This can quickly break down in the real world. Most of us go into CDMOs with already qualified equipment. We have platforms on which we’ve qualified our equipment, too. We don’t know the CPPs until just before PPQ.
This makes the user requirements even more important as living documents. Yes, we’ve qualified our equipment for these large ranges. Now that we have the CPPs, we update the user requirements for the Product User Requirements, perform an overall assessment of the gaps, and, with a risk-based approach, do additional verification activations either before or as part of Process Performance Qualification (PPQ).