Disclaimer: I have had the privilege of being a former colleague of Jayet’s, and hold him in immense regard.
Mastering Safety Risk Management for Medical and In Vitro Devices by Jayet Moon and Arun Mathew is a comprehensive guide that addresses the critical aspects of risk management in medical and in vitro devices. This book is an essential resource for professionals involved in medical device design, production, and post-market phases, providing a structured approach to ensure product safety and regulatory compliance.
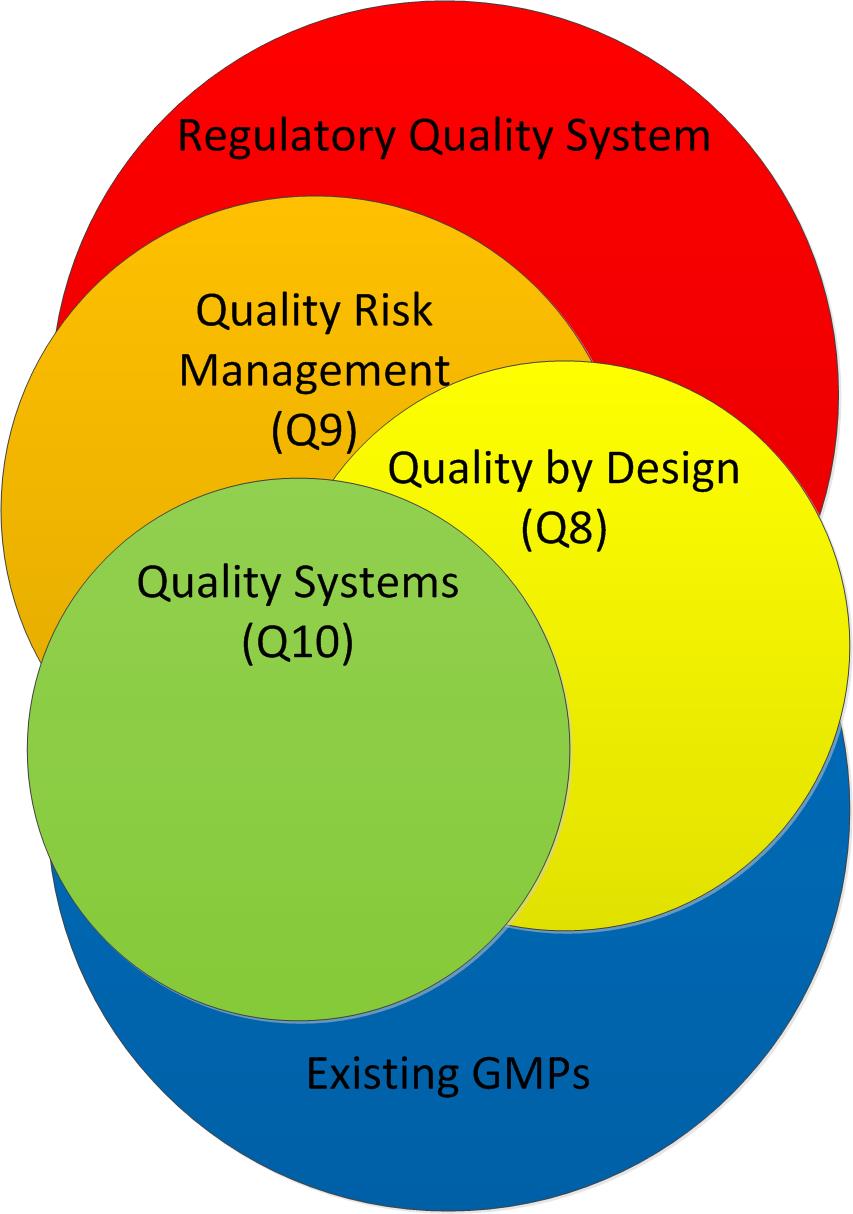
Starting with a solid overview of risk management principles that apply not only to medical devices under ISO13485 but will also teach pharmaceutical folks following ICH Q9 white a bit, this book delivers a heavy dose of knowledge and the benefit of wisdom in applying it.
The book then goes deep into the design assurance process, which is crucial for identifying, understanding, analyzing, and mitigating risks associated with healthcare product design. This foundational approach ensures that practitioners can perform a favorable benefit-risk assessment, which is vital for the safety and efficacy of medical devices.
Strengths
Regulatory Compliance: The authors provide detailed guidance on conforming to major international standards such as ISO 13485:2016, ISO 14971:2019, the European Union Medical Device Regulation (MDR), In Vitro Diagnostic Regulation (IVDR), and the US FDA regulations, including the new FDA Quality Management System Regulation (QMSR).
Risk Management Tools: The book offers a variety of tools and methodologies for effective risk management. These include risk analysis techniques, risk evaluation methods, and risk control measures, which are explained clearly and practically.
Lifecycle Approach: One of the standout features of this book is its lifecycle approach to risk management. It emphasizes that risk management does not end with product design but continues through production and into the post-market phase, ensuring ongoing safety and performance.
The authors, Jayet Moon and Arun Mathew, bring their extensive experience in the field to bear, providing real-world examples and case studies that illustrate the application of risk management principles in various scenarios. This practical approach helps readers to understand how to implement the theoretical concepts discussed in the book. This book is essential for anyone working in medical devices and a good read for other quality life sciences professionals as there is much to draw on here.
An effective program for managing extractables and leachables (E&L) in biotech involves a comprehensive approach that ensures product safety and compliance with regulatory standards. As single-use technologies have become more prevalent in biopharmaceutical manufacturing, leachables from bags, tubing, and other plastic components have become an area of concern. This has led to more rigorous supplier qualification and leachables risk assessment for single-use systems.
Extractables are chemical compounds that can be extracted from materials (like single-use systems, packaging, or manufacturing equipment) under exaggerated conditions such as elevated temperature, extended contact time, or use of strong solvents. They represent a “worst-case” scenario of chemicals potentially migrating into a drug product. Extractables are specific to the tested material and are independent of the drug product.
Leachables are chemical compounds that actually migrate from materials into the drug product under normal conditions of use or storage. They are specific to the combination of the material and the particular drug substance or product, representing the contaminants that may be present in the final drug formulation. Leachables are typically a subset of extractables that migrate under real-world conditions.
The accumulation of extractables and leachabes in a process fluid is governed by thermodynamics (the extent to which the materials would migrate) and kinetic (the rate at which would migrate) factors, as well as the amount of time during which such migration will occur. Higher temperatures increase the migration rate of leachables from the bulk of plastic to the surface in contact with the process stream or formulation.
Key points
Extractables studies are performed on materials using exaggerated conditions.
Leachables studies are performed on the actual drug product under normal conditions.
Extractables represent potential contaminants, while leachables are actual contaminants.
Both are critical for assessing product safety and quality in biotech manufacturing.
Proper evaluation of extractables and leachables is essential for regulatory compliance and ensuring patient safety in biopharmaceutical products.
Program Objectives
Safety Assurance: Ensure that any chemicals leached from materials into the product do not pose a risk to patient safety.
Regulatory Compliance: Meet all relevant regulatory requirements and guidelines.
Quality Control: Maintain the integrity and quality of the biopharmaceutical product.
Regulatory Requirements
Compliance with USP <661> Plastic Packaging Systems and Their Materials of Construction, and USP <381> Elastomeric Closure for Injection
Compliance with USP <87> Biological Reactivity, In Vitro and USP <88> Biological Reactivity, In Vivo
Compliance with European Pharmacopoeia (EP) requirements for materials used in containers, including EP General Chapter 3.1 Materials Used for the Manufacture of Containers and EP 3.2.9 Rubber Closures
Compliance with Japanese Pharmacopoeia (JP) chapter 7.03 Test for Rubber Closures for Aqueous Infusions
Compliance with EU Commission Decision 97/534/EC for Animal derived stearates
Adherence to ICH Q8, Q9, and Q10 guidelines for quality risk management
Leverage ISO 10993-1:2018 Biological evaluation of medical devices
Program Components
Design Space
The starting point should be a review of the supplier’s data. These studies should be performed on materials at the component level under standardized conditions of temperature time, surface, area, etc., so that the data is representative of intended use, including sterilization techniques. Using this data, the end-user can calculate the minimum amount of extractables based on surface area and other conditions. Consider the impact of dilution and clearance over the complete process through risk assessment and then complement with targeted studies.
These studies should be developed based on Quality-by-design principles described in ICH Q8 to gather all the attributes and parameters used to determine a design space. Scientific variables should be identified to set up the Design of Experiment (DoE) for the testing plan.
Risk Assessment
Material Selection: Evaluate materials used for their potential to release harmful substances.
Process Understanding: Understand the process conditions (e.g., temperature, pH, solvents) that might affect the leaching of chemicals.
Risk Prioritization: Prioritize materials and processes based on their risk of contributing harmful leachables. Consider factors like stage of manufacturing, contact time, and proximity to final product.
The risk assessment needs to be within the overall context of process performance and product safety and efficacy. It should not be a separate risk assessment. You will dive deeper with more specific risk questions, but the hazard identification starts at the process level. In evaluating risks the following factors should be considered:
Proximity of the process steps undergoing a change to the final product. Polymeric components in process steps closer to DS or DP will carry a higher risk rating than those in upstream process steps. For example, a bag or filter used for the final filtration of bulk drug substance (BDS) will have a much higher risk rating than components used in upstream process steps since there are no purification steps post-UF/DF.
Storage and processing conditions (e.g., duration of exposure, temperature, pressure, pH extremes, buffer extraction propensity)
The type of process fluid (e.g., purification buffer versus formulated drug substance, presence of solubilizing agents)
Construction materials
Potential adverse events, including synergistic and additive affects
Drug dose, mode, and frequency of administration
Therapeutic necessity
Your risk assessment will drive study design and should consider:
Analytical challenges
Detecting and quantifying trace levels of leachables, which are often present at extremely low concentrations
Developing analytical methods capable of detecting and quantifying a wide range of potential extractables/leachables
Interference from formulation components or degradation products
Determining appropriate extraction conditions:
Selecting solvents and conditions that adequately simulate or exaggerate real-world use conditions
Balancing the need for aggressive extraction (to identify potential leachables) with realistic use conditions
Toxicological assessment
Evaluating the safety impact of identified extractables/leachables, especially for novel compounds
Determining appropriate safety thresholds and analytical evaluation thresholds
Regulatory expectations
Meeting evolving regulatory requirements and expectations, which can vary between regions
Justifying the extent of E&L studies performed based on risk assessment
Unexpected interactions
Leachables causing unexpected effects, such as oxidation of preservatives or formation of protein-leachable adducts
Interactions between leachables and the drug product that were not predicted by extractables studies
Time and resource constraints
E&L studies can be time-consuming and resource-intensive, potentially impacting development timelines
Absorption issues
Adsorption or absorption of drug product components by single-use materials, potentially affecting product stability or efficacy
Stability considerations
Leachables appearing during stability studies that were not identified in initial extractables screening
Changes in leachables profile over time or under different storage conditions
Material variability
Inconsistencies in extractables/leachables profiles between different lots of materials or components
Biopharmaceutical-specific challenges
Potential impact of leachables on sensitive cell lines or biological processes
Interference of leachables with bioassays or analytical methods specific to biologics
Extractables Studies
Objective: Identify potential extractables from materials under exaggerated conditions.
Methodology:
Use a range of solvents that mimic the process fluids.
Apply exaggerated conditions such as elevated temperatures and extended contact times.
Analyze the extracts using techniques like GC-MS, LC-MS, and ICP-MS.
Data Review: Compare supplier-provided extractable data with the intended use to determine the need for specific studies.
Leachables Studies
Objective: Identify and quantify leachables under actual process conditions.
Methodology:
Conduct studies during the development stages and monitor during stability studies.
Use appropriate solvent systems and conditions that mimic the actual process.
Analyze the product for leachables using validated analytical methods.
Toxicological Assessment: Assess the toxicological impact of identified leachables to ensure they are within safe limits.
Migration Studies
Objective: Evaluate the migration of chemicals from materials into the product over time.
Methodology:
Perform studies during the development phase.
Monitor leachables during formal stability studies under normal and accelerated conditions.
Absorption Studies
Objective: Assess the potential for adsorption or absorption of product components.
Methodology:
Conduct studies if stability issues are observed during hold time studies.
Evaluate the impact on product stability and quality.
Stability Studies
Objective: Ensure the stability of the product in contact with materials.
Methodology:
Conduct real-time and accelerated stability studies.
Monitor product quality attributes such as potency, purity, and safety.
Implementation and Validation
Supplier Qualification
Supplier Evaluation: Assess suppliers’ ability to provide materials that meet E&L requirements.
Documentation Review: Ensure suppliers provide comprehensive extractables data and compliance certificates.
In-House Testing
Validation: Validate the findings from supplier data with in-house testing.
Protocol Development: Develop protocols for E&L testing specific to the product and process conditions.
Acceptance Criteria: Establish acceptance criteria based on regulatory guidelines and risk assessments.
Toxicological Assessment and Risk Mitigation
Assess the toxicological impact of identified leachables to ensure they are within safe limits. Perform Risk Mitigation to:
Implement appropriate controls based on risk assessment results
Consider factors like materials selection, process parameters, and analytical testing
Develop strategies to minimize leachables impact on product quality and safety
Continuous Monitoring
Routine Testing: Implement routine testing of leachables during production.
Change Management: Re-evaluate E&L profiles when there are changes in materials, suppliers, or processes.
Training and Education
Staff Training
Awareness: Train staff on the importance of E&L studies and their impact on product safety.
Technical Training: Provide technical training on conducting E&L studies and interpreting results.
Supplier Collaboration
Engagement: Work closely with suppliers to ensure they understand and meet E&L requirements.
Feedback: Provide feedback to suppliers based on study results to improve material quality.
Conclusion
A robust E&L program in biotech is essential for ensuring product safety, regulatory compliance, and maintaining high-quality standards. By implementing a comprehensive approach that includes risk assessment, thorough testing, supplier qualification, continuous monitoring, and staff training, biotech companies can effectively manage the risks associated with extractables and leachables.
I’ve seen my fair share of risk assessments listing data quality or bias as hazards. I tend to think that is pretty sloppy. I especially see this a lot in conversations around AI/ML. Data quality is not a risk. It is a causal factor in the failure or severity.
Data Quality and Data Bias
Data Quality
Data quality refers to how well a dataset meets certain criteria that make it fit for its intended use. The key dimensions of data quality include:
Accuracy – The data correctly represents the real-world entities or events it’s supposed to describe.
Completeness – The dataset contains all the necessary information without missing values.
Consistency – The data is uniform and coherent across different systems or datasets.
Timeliness – The data is up-to-date and available when needed.
Validity – The data conforms to defined business rules and parameters.
Uniqueness – There are no duplicate records in the dataset.
High-quality data is crucial for making informed quality decisions, conducting accurate analyses, and developing reliable AI/ML models. Poor data quality can lead to operational issues, inaccurate insights, and flawed strategies.
Data Bias
Data bias refers to systematic errors or prejudices present in the data that can lead to inaccurate or unfair outcomes, especially in machine learning and AI applications. Some common types of data bias include:
Sampling bias – When the data sample doesn’t accurately represent the entire population.
Selection bias – When certain groups are over- or under-represented in the dataset.
Reporting bias – When the frequency of events in the data doesn’t reflect real-world frequencies.
Measurement bias – When the data collection method systematically skews the results.
Algorithmic bias – When the algorithms or models introduce biases in the results.
Data bias can lead to discriminatory outcomes and produce inaccurate predictions or classifications.
Relationship between Data Quality and Bias
While data quality and bias are distinct concepts, they are closely related:
Poor data quality can introduce or exacerbate biases. For example, incomplete or inaccurate data may disproportionately affect certain groups.
High-quality data doesn’t necessarily mean unbiased data. A dataset can be accurate, complete, and consistent but still contain inherent biases.
Addressing data bias often involves improving certain aspects of data quality, such as completeness and representativeness.
Organizations must implement robust data governance practices to ensure high-quality and unbiased data, regularly assess their data for quality issues and potential biases, and use techniques like data cleansing, resampling, and algorithmic debiasing.
Identifying the Hazards and the Risks
It is critical to remember the difference between a hazard and a risk. Data quality is a causal factor in the hazard, not a harm.
Think of it like a fever. An open wound is a causal factor for the fever, which has a root cause of poor wound hygiene. I can have the factor (the wound), but without the presence of the root cause (poor wound hygiene), the event (fever) would not develop (okay, there may be other root causes in play as well; remember there is never really just one root cause).
Potential Issues of Poor Data Quality and Inadequate Data Governance
The risks associated with poor data quality and inadequate data governance can significantly impact organizations. Here are the key areas where risks can develop:
Decreased Data Quality
Inaccurate, incomplete, or inconsistent data leads to flawed decision-making
Errors in customer information, product details, or financial data can cause operational issues
Poor quality data hinders effective analysis and forecasting
Compliance Failures:
Non-compliance with regulations can result in regulatory actions
Legal complications and reputational damage from failing to meet regulatory requirements
Increased scrutiny from regulatory bodies
Security Breaches
Inadequate data protection increases vulnerability to cyberattacks and data breaches
Financial costs associated with breach remediation, legal fees, and potential fines
Loss of customer trust and long-term reputational damage
Operational Inefficiencies
Time wasted on manual data cleaning and correction
Reduced productivity due to employees working with unreliable data
Inefficient processes resulting from poor data integration or inconsistent data formats
Missed Opportunities
Failure to identify market trends or customer insights due to unreliable data
Missed sales leads or potential customers because of inaccurate contact information
Inability to capitalize on business opportunities due to lack of trustworthy data
Poor Decision-Making
Decisions based on inaccurate or incomplete data leading to suboptimal outcomes, including deviations and product/study impact
Misallocation of resources due to flawed insights from poor quality data
Inability to effectively measure and improve performance
Potential Issues of Data Bias
Data bias presents significant risks across various domains, particularly when integrated into machine learning (ML) and artificial intelligence (AI) systems. These risks can manifest in several ways, impacting both individuals and organizations.
Discrimination and Inequality
Data bias can lead to discriminatory outcomes, systematically disadvantaging certain groups based on race, gender, age, or socioeconomic status. For example:
Judicial Systems: Biased algorithms used in risk assessments for bail and sentencing can result in harsher penalties for people of color compared to their white counterparts, even when controlling for similar circumstances.
Healthcare: AI systems trained on biased medical data may provide suboptimal care recommendations for minority groups, potentially exacerbating health disparities.
Erosion of Trust and Reputation
Organizations that rely on biased data for decision-making risk losing the trust of their customers and stakeholders. This can have severe reputational consequences:
Customer Trust: If customers perceive that an organization’s AI systems are biased, they may lose trust in the brand, leading to a decline in customer loyalty and revenue.
Reputation Damage: High-profile cases of AI bias, such as discriminatory hiring practices or unfair loan approvals, can attract negative media attention and public backlash.
Legal and Regulatory Risks
There are significant legal and regulatory risks associated with data bias:
Compliance Issues: Organizations may face legal challenges and fines if their AI systems violate anti-discrimination laws.
Regulatory Scrutiny: Increasing awareness of AI bias has led to calls for stricter regulations to ensure fairness and accountability in AI systems.
Poor Decision-Making
Biased data can lead to erroneous decisions that negatively impact business operations:
Operational Inefficiencies: AI models trained on biased data may make poor predictions, leading to inefficient resource allocation and operational mishaps.
Financial Losses: Incorrect decisions based on biased data can result in financial losses, such as extending credit to high-risk individuals or mismanaging inventory.
Amplification of Existing Biases
AI systems can perpetuate and even amplify existing biases if not properly managed:
Feedback Loops: Biased AI systems can create feedback loops where biased outcomes reinforce the biased data, leading to increasingly skewed results over time.
Entrenched Inequities: Over time, biased AI systems can entrench societal inequities, making it harder to address underlying issues of discrimination and inequality.
Ethical and Moral Implications
The ethical implications of data bias are profound:
Fairness and Justice: Biased AI systems challenge the principles of fairness and justice, raising moral questions about using such technologies in critical decision-making processes.
Human Rights: There are concerns that biased AI systems could infringe on human rights, particularly in areas like surveillance, law enforcement, and social services.
Perform the Risk Assessment
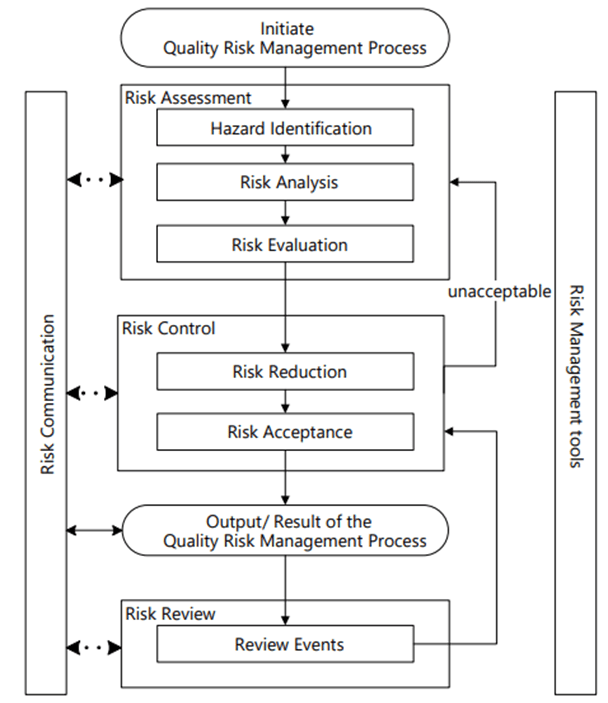
ICH Q9 (r1) Risk Management Process
Risk Management happens at the system/process level, where an AI/ML solution will be used. As appropriate, it drills down to the technology level. Never start with the technology level.
Hazard Identification
It is important to identify product quality hazards that may ultimately lead to patient harm. What is the hazard of that bad decision? What is the hazard of bad quality data? Those are not hazards; they are causes.
Hazard identification, the first step of a risk assessment, begins with a well-defined question defining why the risk assessment is being performed. It helps define the system and the appropriate scope of what will be studied. It addresses the “What might go wrong?” question, including identifying the possible consequences of hazards. The output of the hazard identification step is the identification of the possibilities (i.e., hazards) that the risk event (e.g., impact to product quality) happens.
The risk question takes the form of “What is the risk of using AI/ML solution for <Process/System> to <purpose of AI/MIL solution.” For example, “What is the risk of using AI/ML to identify deviation recurrence and help prioritize CAPAs?” or “What is the risk of using AI/ML to monitor real-time continuous manufacturing to determine the need to evaluate for a potential diversion?”
We can now identify the specific failure modes associated with AI/ML. This may involve deeep dive risk assessments. A failure mode is the specific way a failure occurs. So in this case, the specific way that bad data or bad decision making can happen. Multiple failure modes can, and usually do, lead to the same hazardous situation.
Make sure you drill down on failure causes. If more than 5 potential causes can be identified for a proposed failure mode, it is too broad and probably written at a high level in the process or item being risk assessed. It should be broken down into several specific failure modes with fewer potential causes and more manageable.
Start with an outline of how the process works and a description of the AI/ML (special technology) used in the process. Then, interrogate the following for potential failure modes:
The steps in the process or item under study in which AI/ML interventions occur;
The process/procedure documentation for example, master batch records, SOPs, protocols, etc.
Current and proposed process/procedure in sufficient detail to facilitate failure mode identification;
One of the topics I’m passionate about is exploring the changing landscape of quality management and the challenges we face. The solutions that worked in the past decade won’t be as effective in our current era, marked by post-globalization, capital rationalization, spatial dispersion, shrinking workforces, and an increasing reliance on automation. This transformation calls for a new perspective on quality management, as traditional instincts and strategies may no longer be sufficient. The nature of opportunity and risk has fundamentally changed, and in order to thrive, we need to adapt our approach.
The New Rules of Engagement
In this era of volatility, several key trends are reshaping the business environment:
Post-Globalization: The shift towards localized operations and supply chains.
Capital Rationalization: More stringent allocation of financial resources. This is a huge trend in biotech.
Spatial Dispersion: Decentralized workforces and operations.
Shrinking Workforces: Reduced human resources due to demographic changes.
Dependence on Automation: Increased reliance on technologies like AI, ML, and RPA.
We need to reevaluate how we approach quality management in light of these trends.
Prediction: Anticipating the Future
In a volatile environment, it is crucial to predict and anticipate disruptions. Quality management must shift from being reactive to proactive. This involves:
Advanced Analytics: Utilizing data analytics to anticipate quality issues before they emerge. This necessitates a strong data foundation and the capability to analyze both structured and unstructured data.
Scenario Planning: Developing multiple scenarios to anticipate potential disruptions and their impacts on quality aids in making well-informed strategic decisions and preparing for various contingencies.
Adaptability: Embracing Change
Adaptability is crucial in a constantly changing world. Quality management systems need to be flexible and responsive to new challenges.
Agile Methodologies: Implementing agile practices to allow for quick adjustments to processes and workflows, fostering a culture of experimentation, and learning from failures.
Virtualization of Work: Adapting quality processes to support remote and hybrid work environments involves re-evaluating governance models and ensuring that quality standards are maintained regardless of the location of work.
Resilience: Building Robust Systems
Resilience ensures that organizations can withstand and recover from disruptions. This capability is built on strong foundations:
Robust Systems: Developing systems that can operate effectively under stress. This includes ensuring that automated processes are reliable and that there are contingencies for system failures.
Organizational Culture: Fostering a culture that values resilience and continuous improvement ensures that employees are prepared to handle disruptions and contribute to the organization’s long-term success.
Implementing the New Quality Paradigm
To effectively implement these principles, organizations should consider the following steps:
Assess the Current State: Conduct a comprehensive assessment of existing quality processes, identifying areas for improvement and potential vulnerabilities.
Set Clear Objectives: Establish clear, measurable objectives that align with the principles of prediction, adaptability, and resilience.
Develop a Phased Approach: Implement changes gradually, with clear milestones and measurable outcomes to ensure smooth transitions.
Engage Stakeholders: Involve all relevant stakeholders in the transformation process to ensure alignment and buy-in.
Monitor Progress: Continuously monitor progress against predefined objectives and make adjustments as necessary to stay on track.
Invest in Training: Provide employees with the necessary training and development opportunities to adapt to new technologies and processes.
Conclusion
It is important to change our mindset and strategy. Embracing the principles of prediction, adaptability, and resilience can help organizations navigate the complexities of a volatile environment and position themselves for long-term success. Going forward, it is essential to stay vigilant, flexible, and proactive in our approach to quality management. We must ensure that we not only meet but exceed stakeholder expectations in this rapidly changing world.
Too often, I see folks in pharma focus on 21 CFR Chapter 1, or at best all three chapters, maybe know the guidances and pay attention to little else. Unfortunately, that approach will often get one in trouble.
Section 711 of the Food and Drug Administration Safety and Innovation Act (FDASIA) amended the Federal Food, Drug, and Cosmetic Act (FD&C Act) to enhance the safety and quality of the drug supply chain. Specifically Section 711 amends Section 501(a)(2)(B) of the FD&C Act by adding the following sentence:
“For purposes of paragraph (a)(2)(B), the term ‘current good manufacturing practice’ includes the implementation of oversight and controls over the manufacture of drugs to ensure quality, including managing the risk of and establishing the safety of raw materials, materials used in the manufacturing of drugs, and finished drug products.”
This amendment clarifies that current good manufacturing practice (CGMP) requirements for drugs include:
Implementing oversight and controls over the entire manufacturing process to ensure quality.
Managing the risks related to raw materials, other materials used in manufacturing, and finished drug products to establish their safety.
In essence, Section 711 expands the FDA’s CGMP authority to explicitly cover supply chain management and drug manufacturers’ oversight of their suppliers and contract manufacturing operations. It also allows the FDA to enforce supply chain control requirements during inspections.
The legislative history shows that Congress intended to significantly expand the FDA’s authority over the increasingly global drug supply chain through this provision. It allows the FDA to scrutinize how manufacturers select, qualify, and oversee suppliers of raw materials and contract manufacturers to ensure drug quality and safety.
Please note that the FDA gets this expanded authority without revising 21CFR. That’s how it works; Congress can do that. Will we eventually see some 21 CFR updates? I have no idea.
But what this does mean is that the FDA has the authority to:
Inspect risk management for GMPs, and assume you have it. What does good risk management look like? The agency has adopted ICH Q9(r1) as guidance, so start there.
Inspect your supplier management, which includes qualifying and overseeing suppliers and contract manufacturers.
I’ve started to receive regulatory intelligence that this is coming up in inspections. Expect to be asked for the risk management evidence and for supplier qualification and oversight evidence.



{kind=link}