The relationship between sponsors and contract organizations has evolved far beyond simple transactional exchanges. Digital infrastructure has become the cornerstone of trust, transparency, and operational excellence.
The trust equation is fundamentally changing due to the way our supply chains are being challenged.. Traditional quality agreements often functioned as static documents—comprehensive but disconnected from day-to-day operations. Today’s most successful partnerships are built on dynamic, digitally-enabled frameworks that provide real-time visibility into performance, compliance, and risk management.
Regulatory agencies are increasingly scrutinizing the effectiveness of sponsor oversight programs. The FDA’s emphasis on data integrity, combined with EMA’s evolving computerized systems requirements, means that sponsors can no longer rely on periodic audits and static documentation to demonstrate control over their outsourced activities.
Quality Agreements as Digital Trust Frameworks
The modern quality agreement must evolve from a compliance document to a digital trust framework. This transformation requires reimagining three fundamental components:
Dynamic Risk Assessment Integration
Traditional quality agreements categorize suppliers into static risk tiers (for example Category 1, 2, 2.5, or 3 based on material/service risk). Digital frameworks enable continuous risk profiling that adapts based on real-time performance data.
Integrate supplier performance metrics directly into your quality management system. When a Category 2 supplier’s on-time delivery drops below threshold or quality metrics deteriorate, the system should automatically trigger enhanced monitoring protocols without waiting for the next periodic review.
Automated Change Control Workflows
One of the most contentious areas in sponsor-CxO relationships involves change notifications and approvals. Digital infrastructure can transform this friction point into a competitive advantage.
The SMART approach to change control:
Standardized digital templates for change notifications
Machine-readable impact assessments
Automated routing based on change significance
Real-time status tracking for all stakeholders
Traceable decision logs with electronic signatures
Quality agreement language to include: “All change notifications shall be submitted through the designated digital platform within [X] business days of identification, with automated acknowledgment and preliminary impact assessment provided within [Y] hours.”
Transparent Performance Dashboards
The most innovative CxOs are moving beyond quarterly business reviews to continuous performance visibility. Quality agreements should build upon real-time access to key performance indicators (KPIs) that matter most to patient safety and product quality.
Examples of Essential KPIs for digital dashboards:
Batch disposition times and approval rates
Deviation investigation cycle times
CAPA effectiveness metrics
Environmental monitoring excursions and response times
Supplier change notification compliance rates
Communication Architecture for Transparency
Effective communication in pharmaceutical partnerships requires architectural thinking, not just protocol definition. The most successful CxO-sponsor relationships are built on what I call the “Three-Layer Communication Stack” which builds a rhythm of communication:
Layer 1: Operational Communication (Real-Time)
Purpose: Day-to-day coordination and issue resolution
Tools: Integrated messaging within quality management systems, automated alerts, mobile notifications
Quality agreement requirement: “Operational communications shall be conducted through validated, audit-trailed platforms with 24/7 availability and guaranteed delivery confirmation.”
Every quality agreement should include a subsidiary Communication Plan that addresses:
Stakeholder Matrix: Who needs what information, when, and in what format
Escalation Protocols: Clear triggers for moving issues up the communication stack
Performance Metrics: How communication effectiveness will be measured and improved
Technology Requirements: Specified platforms, security requirements, and access controls
Contingency Procedures: Alternative communication methods for system failures or emergencies
Include communication effectiveness as a measurable element in your supplier scorecards. Track metrics like response time to quality notifications, accuracy of status reporting, and proactive problem identification.
Data Governance as a Competitive Differentiator
Data integrity is more than just ensuring ALCOA+—it’s about creating a competitive moat through superior data governance. The organizations that master data sharing, analysis, and decision-making will dominate the next decade of pharmaceutical manufacturing and development.
The Modern Data Governance Framework
Data Architecture Definition
Your quality agreement must specify not just what data will be shared, but how it will be structured, validated, and integrated:
Master data management: Consistent product codes, batch numbering, and material identifiers across all systems
Data quality standards: Validation rules, completeness requirements, and accuracy thresholds
Integration protocols: APIs, data formats, and synchronization frequencies
With increasing regulatory focus on cybersecurity, your data governance plan must address:
Role-based access controls: Granular permissions based on job function and business need
Data classification: Confidentiality levels and handling requirements
Audit logging: Comprehensive tracking of data access, modification, and sharing
Analytics and Intelligence
The real competitive advantage comes from turning shared data into actionable insights:
Predictive analytics: Early warning systems for quality trends and supply chain disruptions
Benchmark reporting: Anonymous industry comparisons to identify improvement opportunities
Root cause analysis: Automated correlation of events across multiple systems and suppliers
The Data Governance Subsidiary Agreement
Consider creating a separate Data Governance Agreement that complements your quality agreement with specific sections covering data sharing objectives, technical architecture, governance oversight, and compliance requirements.
Veeva Summit
Next week I’ll be discussing this topic at the Veeva Summit, where I will bring some organizational learnings on to embrace digital infrastructure as a trust-building mechanism will forge stronger partnerships, achieve superior quality outcomes, and ultimately deliver better patient experiences.
The pharmaceutical industry has operated for over a decade under the comfortable assumption that GAMP 5’s risk-based guidance for system requirements represented industry best practice—helpful, comprehensive, but ultimately voluntary. Section 6 of the draft Annex 11 moves many things from recommended to mandated. What GAMP 5 suggested as scalable guidance, Annex 11 codifies as enforceable regulation. For computer system validation professionals, this isn’t just an update—it’s a fundamental shift from “how we should do it” to “how we must do it.”
This transformation carries profound implications that extend far beyond documentation requirements. Section 6 represents the regulatory codification of modern system engineering practices, forcing organizations to abandon the shortcuts, compromises, and “good enough” approaches that have persisted despite GAMP 5’s guidance. More significantly, it establishes system requirements as the immutable foundation of validation rather than merely an input to the process.
For CSV experts who have spent years evangelizing GAMP 5 principles within organizations that treated requirements as optional documentation, Section 6 provides regulatory teeth that will finally compel comprehensive implementation. However, it also raises the stakes dramatically—what was once best practice guidance subject to interpretation becomes regulatory obligation subject to inspection.
The Mandatory Transformation: From Guidance to Regulation
6.1: GMP Functionality—The End of Requirements Optionality
The opening requirement of Section 6 eliminates any ambiguity about system requirements documentation: “A regulated user should establish and approve a set of system requirements (e.g. a User Requirements Specification, URS), which accurately describe the functionality the regulated user has automated and is relying on when performing GMP activities.”
This language transforms what GAMP 5 positioned as risk-based guidance into regulatory mandate. The phrase “should establish and approve” in regulatory context carries the force of “must“—there is no longer discretion about whether to document system requirements. Every computerized system touching GMP activities requires formal requirements documentation, regardless of system complexity, development approach, or organizational preference.
The scope is deliberately comprehensive, explicitly covering “whether a system is developed in-house, is a commercial off-the-shelf product, or is provided as-a-service” and “independently on whether it is developed following a linear or iterative software development process.” This eliminates common industry escapes: cloud services can’t claim exemption because they’re external; agile development can’t avoid documentation because it’s iterative; COTS systems can’t rely solely on vendor documentation because they’re pre-built.
The requirement for accuracy in describing “functionality the regulated user has automated and is relying on” establishes a direct link between system capabilities and GMP dependencies. Organizations must explicitly identify and document what GMP activities depend on system functionality, creating traceability between business processes and technical capabilities that many current validation approaches lack.
Major Strike Against the Concept of “Indirect”
The new draft Annex 11 explicitly broadens the scope of requirements for user requirements specifications (URS) and validation to cover all computerized systems with GMP relevance—not just those with direct product or decision-making impact, but also indirect GMP systems. This means systems that play a supporting or enabling role in GMP activities (such as underlying IT infrastructure, databases, cloud services, SaaS platforms, integrated interfaces, and any outsourced or vendor-managed digital environments) are fully in scope.
Section 6 of the draft states that user requirements must “accurately describe the functionality the regulated user has automated and is relying on when performing GMP activities,” with no exemption or narrower definition for indirect systems. It emphasizes that this principle applies “regardless of whether a system is developed in-house, is a commercial off-the-shelf product, or is provided as-a-service, and independently of whether it is developed following a linear or iterative software development process.” The regulated user is responsible for approving, controlling, and maintaining these requirements over the system’s lifecycle—even if the system is managed by a third party or only indirectly involved in GMP data or decision workflows.
Importantly, the language and supporting commentaries make it clear that traceability of user requirements throughout the lifecycle is mandatory for all systems with GMP impact—direct or indirect. There is no explicit exemption in the draft for indirect GMP systems. Regulatory and industry analyses confirm that the burden of documented, risk-assessed, and lifecycle-maintained user requirements sits equally with indirect systems as with direct ones, as long as they play a role in assuring product quality, patient safety, or data integrity.
In practice, this means organizations must extend their URS, specification, and validation controls to any computerized system that through integration, support, or data processing could influence GMP compliance. The regulated company remains responsible for oversight, traceability, and quality management of those systems, whether or not they are operated by a vendor or IT provider. This is a significant expansion from previous regulatory expectations and must be factored into computerized system inventories, risk assessments, and validation strategies going forward.
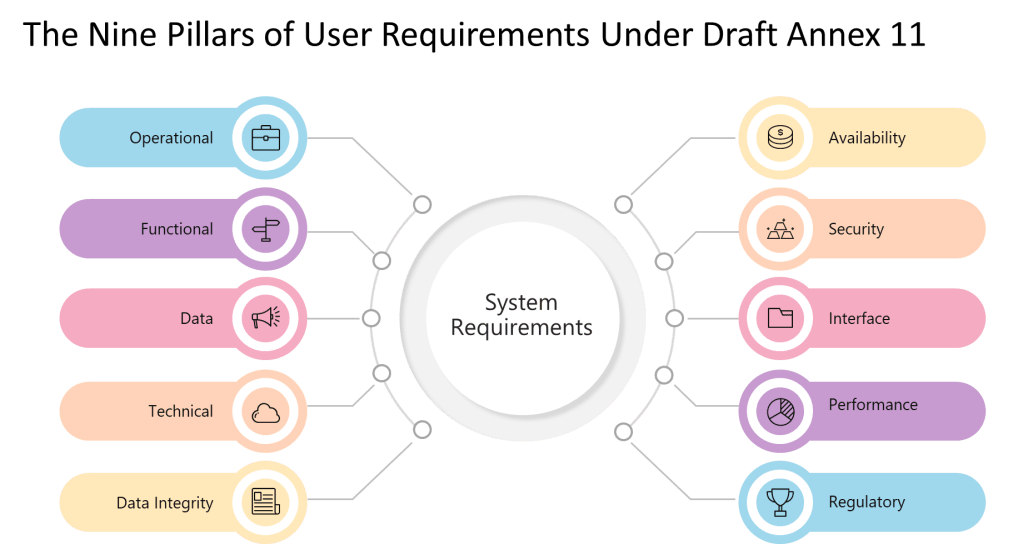
9 Pillars of a User Requirements
Pillar
Description
Practical Examples
Operational
Requirements describing how users will operate the system for GMP tasks.
Workflow steps, user roles, batch record creation.
Functional
Features and functions the system must perform to support GMP processes.
Explicit requirements imposed by GMP regulations and standards.
Part 11/Annex 11 compliance, data retention, auditability.
6.2: Extent and Detail—Risk-Based Rigor, Not Risk-Based Avoidance
Section 6.2 appears to maintain GAMP 5’s risk-based philosophy by requiring that “extent and detail of defined requirements should be commensurate with the risk, complexity and novelty of a system.” However, the subsequent specifications reveal a much more prescriptive approach than traditional risk-based frameworks.
The requirement that descriptions be “sufficient to support subsequent risk analysis, specification, design, purchase, configuration, qualification and validation” establishes requirements documentation as the foundation for the entire system lifecycle. This moves beyond GAMP 5’s emphasis on requirements as input to validation toward positioning requirements as the definitive specification against which all downstream activities are measured.
The explicit enumeration of requirement types—”operational, functional, data integrity, technical, interface, performance, availability, security, and regulatory requirements”—represents a significant departure from GAMP 5’s more flexible categorization. Where GAMP 5 allows organizations to define requirement categories based on system characteristics and business needs, Annex 11 mandates coverage of nine specific areas regardless of system type or risk level.
This prescriptive approach reflects regulatory recognition that organizations have historically used “risk-based” as justification for inadequate requirements documentation. By specifying minimum coverage areas, Section 6 establishes a floor below which requirements documentation cannot fall, regardless of risk assessment outcomes.
The inclusion of “process maps and data flow diagrams” as recommended content acknowledges the reality that modern pharmaceutical operations involve complex, interconnected systems where understanding data flows and process dependencies is essential for effective validation. This requirement will force organizations to develop system-level understanding rather than treating validation as isolated technical testing.
6.3: Ownership—User Accountability in the Cloud Era
Perhaps the most significant departure from traditional industry practice, Section 6.3 addresses the growing trend toward cloud services and vendor-supplied systems by establishing unambiguous user accountability for requirements documentation. The requirement that “the regulated user should take ownership of the document covering the implemented version of the system and formally approve and control it” eliminates common practices where organizations rely entirely on vendor-provided documentation.
This requirement acknowledges that vendor-supplied requirements specifications rarely align perfectly with specific organizational needs, GMP processes, or regulatory expectations. While vendors may provide generic requirements documentation suitable for broad market applications, pharmaceutical organizations must customize, supplement, and formally adopt these requirements to reflect their specific implementation and GMP dependencies.
The language “carefully review and approve the document and consider whether the system fulfils GMP requirements and company processes as is, or whether it should be configured or customised” requires active evaluation rather than passive acceptance. Organizations cannot simply accept vendor documentation as sufficient—they must demonstrate that they have evaluated system capabilities against their specific GMP needs and either confirmed alignment or documented necessary modifications.
This ownership requirement will prove challenging for organizations using large cloud platforms or SaaS solutions where vendors resist customization of standard documentation. However, the regulatory expectation is clear: pharmaceutical companies cannot outsource responsibility for demonstrating that system capabilities meet their specific GMP requirements.
6.4: Update—Living Documentation, Not Static Archives
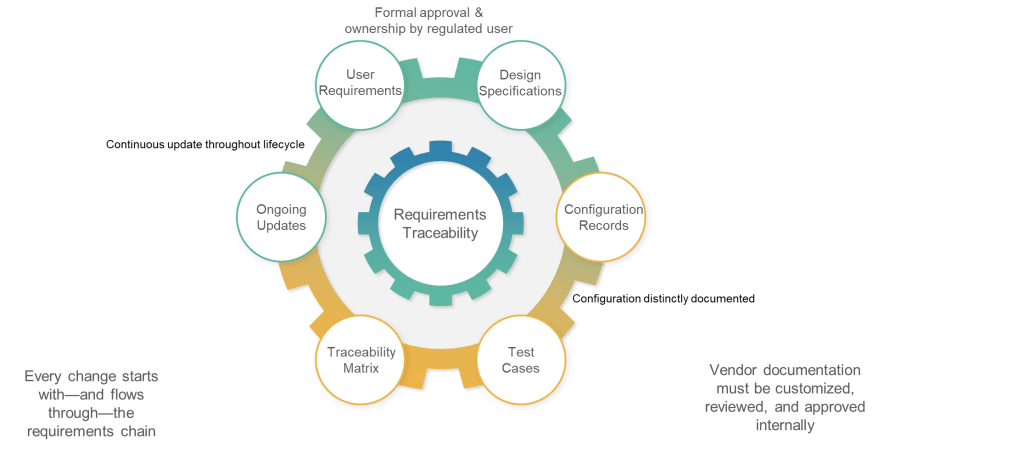
Section 6.4 addresses one of the most persistent failures in current validation practice: requirements documentation that becomes obsolete immediately after initial validation. The requirement that “requirements should be updated and maintained throughout the lifecycle of a system” and that “updated requirements should form the very basis for qualification and validation” establishes requirements as living documentation rather than historical artifacts.
This approach reflects the reality that modern computerized systems undergo continuous change through software updates, configuration modifications, hardware refreshes, and process improvements. Traditional validation approaches that treat requirements as fixed specifications become increasingly disconnected from operational reality as systems evolve.
The phrase “form the very basis for qualification and validation” positions requirements documentation as the definitive specification against which system performance is measured throughout the lifecycle. This means that any system change must be evaluated against current requirements, and any requirements change must trigger appropriate validation activities.
This requirement will force organizations to establish requirements management processes that rival those used in traditional software development organizations. Requirements changes must be controlled, evaluated for impact, and reflected in validation documentation—capabilities that many pharmaceutical organizations currently lack.
6.5: Traceability—Engineering Discipline for Validation
The traceability requirement in Section 6.5 codifies what GAMP 5 has long recommended: “Documented traceability between individual requirements, underlaying design specifications and corresponding qualification and validation test cases should be established and maintained.” However, the regulatory context transforms this from validation best practice to compliance obligation.
The emphasis on “effective tools to capture and hold requirements and facilitate the traceability” acknowledges that manual traceability management becomes impractical for complex systems with hundreds or thousands of requirements. This requirement will drive adoption of requirements management tools and validation platforms that can maintain automated traceability throughout the system lifecycle.
Traceability serves multiple purposes in the validation context: ensuring comprehensive test coverage, supporting impact assessment for changes, and providing evidence of validation completeness. Section 6 positions traceability as fundamental validation infrastructure rather than optional documentation enhancement.
For organizations accustomed to simplified validation approaches where test cases are developed independently of detailed requirements, this traceability requirement represents a significant process change requiring tool investment and training.
6.6: Configuration—Separating Standard from Custom
The final subsection addresses configuration management by requiring clear documentation of “what functionality, if any, is modified or added by configuration of a system.” This requirement recognizes that most modern pharmaceutical systems involve significant configuration rather than custom development, and that configuration decisions have direct impact on validation scope and approaches.
The distinction between standard system functionality and configured functionality is crucial for validation planning. Standard functionality may be covered by vendor testing and certification, while configured functionality requires user validation. Section 6 requires this distinction to be explicit and documented.
The requirement for “controlled configuration specification” separate from requirements documentation reflects recognition that configuration details require different management approaches than functional requirements. Configuration specifications must reflect the actual system implementation rather than desired capabilities.
Comparison with GAMP 5: Evolution Becomes Revolution
Philosophical Alignment with Practical Divergence
Section 6 maintains GAMP 5’s fundamental philosophy—risk-based validation supported by comprehensive requirements documentation—while dramatically changing implementation expectations. Both frameworks emphasize user ownership of requirements, lifecycle management, and traceability as essential validation elements. However, the regulatory context of Annex 11 transforms voluntary guidance into enforceable obligation.
GAMP 5’s flexibility in requirements categorization and documentation approaches reflects its role as guidance suitable for diverse organizational contexts and system types. Section 6’s prescriptive approach reflects regulatory recognition that flexibility has often been interpreted as optionality, leading to inadequate requirements documentation that fails to support effective validation.
The risk-based approach remains central to both frameworks, but Section 6 establishes minimum standards that apply regardless of risk assessment outcomes. While GAMP 5 might suggest that low-risk systems require minimal requirements documentation, Section 6 mandates coverage of nine requirement areas for all GMP systems.
Documentation Structure and Content
GAMP 5’s traditional document hierarchy—URS, Functional Specification, Design Specification—becomes more fluid under Section 6, which focuses on ensuring comprehensive coverage rather than prescribing specific document structures. This reflects recognition that modern development approaches, including agile and DevOps practices, may not align with traditional waterfall documentation models.
However, Section 6’s explicit enumeration of requirement types provides more prescriptive guidance than GAMP 5’s flexible approach. Where GAMP 5 might allow organizations to define requirement categories based on system characteristics, Section 6 mandates coverage of operational, functional, data integrity, technical, interface, performance, availability, security, and regulatory requirements.
The emphasis on process maps, data flow diagrams, and use cases reflects modern system complexity where understanding interactions and dependencies is essential for effective validation. GAMP 5 recommends these approaches for complex systems; Section 6 suggests their use “where relevant” for all systems.
Vendor and Service Provider Management
Both frameworks emphasize user responsibility for requirements even when vendors provide initial documentation. However, Section 6 uses stronger language about user ownership and control, reflecting increased regulatory concern about organizations that delegate requirements definition to vendors without adequate oversight.
GAMP 5’s guidance on supplier assessment and leveraging vendor documentation remains relevant under Section 6, but the regulatory requirement for user ownership and approval creates higher barriers for simply accepting vendor-provided documentation as sufficient.
Implementation Challenges for CSV Professionals
Organizational Capability Development
Most pharmaceutical organizations will require significant capability development to meet Section 6 requirements effectively. Traditional validation teams focused on testing and documentation must develop requirements engineering capabilities comparable to those found in software development organizations.
This transformation requires investment in requirements management tools, training for validation professionals, and establishment of requirements governance processes. Organizations must develop capabilities for requirements elicitation, analysis, specification, validation, and change management throughout the system lifecycle.
The traceability requirement particularly challenges organizations accustomed to informal relationships between requirements and test cases. Automated traceability management requires tool investments and process changes that many validation teams are unprepared to implement.
Integration with Existing Validation Approaches
Section 6 requirements must be integrated with existing validation methodologies and documentation structures. Organizations following traditional IQ/OQ/PQ approaches must ensure that requirements documentation supports and guides qualification activities rather than existing as parallel documentation.
The requirement for requirements to “form the very basis for qualification and validation” means that test cases must be explicitly derived from and traceable to documented requirements. This may require significant changes to existing qualification protocols and test scripts.
Organizations using risk-based validation approaches aligned with GAMP 5 guidance will find philosophical alignment with Section 6 but must adapt to more prescriptive requirements for documentation content and structure.
Technology and Tool Requirements
Effective implementation of Section 6 requirements typically requires requirements management tools capable of supporting specification, traceability, change control, and lifecycle management. Many pharmaceutical validation teams currently lack access to such tools or experience in their use.
Tool selection must consider integration with existing validation platforms, support for regulated environments, and capabilities for automated traceability maintenance. Organizations may need to invest in new validation platforms or significantly upgrade existing capabilities.
The emphasis on maintaining requirements throughout the system lifecycle requires tools that support ongoing requirements management rather than just initial documentation. This may conflict with validation approaches that treat requirements as static inputs to qualification activities.
Strategic Implications for the Industry
Convergence of Software Engineering and Pharmaceutical Validation
Section 6 represents convergence between pharmaceutical validation practices and mainstream software engineering approaches. Requirements engineering, long established in software development, becomes mandatory for pharmaceutical computerized systems regardless of development approach or vendor involvement.
This convergence benefits the industry by leveraging proven practices from software engineering while maintaining the rigor and documentation requirements essential for regulated environments. However, it requires pharmaceutical organizations to develop capabilities traditionally associated with software development rather than manufacturing and quality assurance.
The result should be more robust validation practices better aligned with modern system development approaches and capable of supporting the complex, interconnected systems that characterize contemporary pharmaceutical operations.
Vendor Relationship Evolution
Section 6 requirements will reshape relationships between pharmaceutical companies and system vendors. The requirement for user ownership of requirements documentation means that vendors must support more sophisticated requirements management processes rather than simply providing generic specifications.
Vendors that can demonstrate alignment with Section 6 requirements through comprehensive documentation, traceability tools, and support for user customization will gain competitive advantages. Those that resist pharmaceutical-specific requirements management approaches may find their market opportunities limited.
The emphasis on configuration management will drive vendors to provide clearer distinctions between standard functionality and customer-specific configurations, supporting more effective validation planning and execution.
The Regulatory Codification of Modern Validation
Section 6 of the draft Annex 11 represents the regulatory codification of modern computerized system validation practices. What GAMP 5 recommended through guidance, Annex 11 mandates through regulation. What was optional becomes obligatory; what was flexible becomes prescriptive; what was best practice becomes compliance requirement.
For CSV professionals, Section 6 provides regulatory support for comprehensive validation approaches while raising the stakes for inadequate implementation. Organizations that have struggled to implement effective requirements management now face regulatory obligation rather than just professional guidance.
The transformation from guidance to regulation eliminates organizational discretion about requirements documentation quality and comprehensiveness. While risk-based approaches remain valid for scaling validation effort, minimum standards now apply regardless of risk assessment outcomes.
Success under Section 6 requires pharmaceutical organizations to embrace software engineering practices for requirements management while maintaining the documentation rigor and process control essential for regulated environments. This convergence benefits the industry by improving validation effectiveness while ensuring compliance with evolving regulatory expectations.
The industry faces a choice: proactively develop capabilities to meet Section 6 requirements or reactively respond to inspection findings and enforcement actions. For organizations serious about digital transformation and validation excellence, Section 6 provides a roadmap for regulatory-compliant modernization of validation practices.
Requirement Area
Draft Annex 11 Section 6
GAMP 5 Requirements
Key Implementation Considerations
System Requirements Documentation
Mandatory – Must establish and approve system requirements (URS)
Recommended – URS should be developed based on system category and complexity
Organizations must document requirements for ALL GMP systems, regardless of size or complexity
Risk-Based Approach
Extent and detail must be commensurate with risk, complexity, and novelty
Risk-based approach fundamental – validation effort scaled to risk
Risk assessment determines documentation detail but cannot eliminate requirement categories
Functional Requirements
Must include 9 specific requirement types: operational, functional, data integrity, technical, interface, performance, availability, security, regulatory
Functional requirements should be SMART (Specific, Measurable, Achievable, Realistic, Testable)
All 9 areas must be addressed; risk determines depth, not coverage
Traceability Requirements
Documented traceability between requirements, design specs, and test cases required
Traceability matrix recommended – requirements linked through design to testing
Requires investment in traceability tools and processes for complex systems
Requirement Ownership
Regulated user must take ownership even if vendor provides initial requirements
User ownership emphasized, even for purchased systems
Cannot simply accept vendor documentation; must customize and formally approve
Lifecycle Management
Requirements must be updated and maintained throughout system lifecycle
Requirements managed through change control throughout lifecycle
Requires ongoing requirements management process, not just initial documentation
Configuration Management
Configuration options must be described in requirements; chosen configuration documented in controlled spec
Configuration specifications separate from URS
Must clearly distinguish between standard functionality and configured features
Vendor-Supplied Requirements
Vendor requirements must be reviewed, approved, and owned by regulated user
Supplier assessment required – leverage supplier documentation where appropriate
Higher burden on users to customize vendor documentation for specific GMP needs
Validation Basis
Updated requirements must form basis for system qualification and validation
Requirements drive validation strategy and testing scope
Requirements become definitive specification against which system performance is measured
The pharmaceutical industry’s approach to supplier management has operated on a comfortable fiction for decades: as long as you had a signed contract and conducted an annual questionnaire review, regulatory responsibility somehow transferred to your vendors. That cozy delusion is shattered to a surprising degree in the new Section 7 of the draft Annex 11, which reads like a regulatory autopsy of every failed outsourcing arrangement that ever derailed a drug approval or triggered a warning letter.
If you’ve been following my earlier breakdowns of the draft Annex 11 overhaul, you know this isn’t incremental tinkering. The regulators are systematically dismantling every assumption about digital compliance that pharmaceutical companies have built their strategies around. Nowhere is this more evident than in Section 7, which transforms supplier management from a procurement afterthought into the backbone of GxP data integrity.
The new requirements don’t just raise the bar—they relocate it to a different planet entirely. Organizations that treat vendor management as a checkbox exercise are about to discover that their carefully constructed compliance programs have been built on quicksand. The draft makes one thing crystal clear: you cannot outsource responsibility, only tasks. Every cloud service, every SaaS platform, every IT support contract becomes a direct extension of your quality management system, subject to the same scrutiny as your in-house operations.
This represents more than regulatory updating. Section 7 acknowledges that modern pharmaceutical operations depend fundamentally on external providers—from cloud infrastructure underpinning LIMS systems to SaaS platforms managing clinical data to third-party IT support maintaining manufacturing execution systems. The old model of “trust but check-in once a year” has been replaced with “prove it, continuously, or prepare for the consequences.”
The Regulatory Context: Why Section 7 Emerged
The current Annex 11, published in 2011, addresses suppliers through a handful of brief clauses that seem almost quaint in retrospect. Section 3 requires “formal agreements” with “clear statements of responsibilities” and suggests that “competence and reliability” should guide supplier selection. The audit requirement appears as a single sentence recommending risk-based assessment. That’s it. Five sentences to govern relationships that now determine whether pharmaceutical companies can manufacture products, release batches, or maintain regulatory compliance.
As digital transformation accelerated throughout the pharmaceutical industry, the guidance became increasingly outdated. Organizations moved core GMP functions to cloud platforms, implemented SaaS quality management systems, and relied increasingly on external IT support—all while operating under regulatory guidance designed for a world where “computerized systems” meant locally installed software running on company-owned hardware.
The regulatory wake-up call came through a series of high-profile data integrity failures, cybersecurity breaches, and compliance failures that traced directly to inadequate supplier oversight. Warning letters began citing “failure to ensure that service providers meet applicable requirements” and “inadequate oversight of computerized system suppliers.” Inspection findings revealed organizations that couldn’t explain how their cloud providers managed data, couldn’t access their audit trails, and couldn’t demonstrate control over systems essential to product quality.
Section 7 represents the regulatory response to this systemic failure. The draft Annex 11 approaches supplier management with the same rigor previously reserved for manufacturing processes, recognizing that in digitized pharmaceutical operations, the distinction between internal and external systems has become largely meaningless from a compliance perspective.
Dissecting Section 7: The Five Subsections That Change Everything
7.1 Responsibility: The Death of Liability Transfer
The opening salvo of Section 7 eliminates any ambiguity about accountability: “When a regulated user is relying on a vendor’s qualification of a system used in GMP activities, a service provider, or an internal IT department’s qualification and/or operation of such system, this does not change the requirements put forth in this document. The regulated user remains fully responsible for these activities based on the risk they constitute on product quality, patient safety and data integrity.”
TThis language represents a fundamental shift from the permissive approach of the 2011 version. Organizations can no longer treat outsourcing as risk transfer. Whether you’re using Amazon Web Services to host your quality management system, Microsoft Azure to run your clinical data platform, or a specialized pharmaceutical SaaS provider for batch record management, you remain fully accountable for ensuring those systems meet every requirement specified in Annex 11.
The practical implications are staggering. Organizations that have structured their compliance programs around the assumption that “the vendor handles validation” must completely reconceptualize their approach. Cloud service providers don’t become exempt from GxP requirements simply because they’re external entities. SaaS platforms can’t claim immunity from data integrity standards because they serve multiple industries. Every system that touches GMP activities becomes subject to the same validation, documentation, and control requirements regardless of where it operates or who owns the infrastructure.
This requirement also extends to internal IT departments, acknowledging that many pharmaceutical organizations have tried to create an artificial separation between quality functions and IT support. The draft eliminates this distinction, making clear that IT departments supporting GMP activities are subject to the exact oversight requirements as external service providers.
The responsibility clause creates particular challenges for organizations using multi-tenant SaaS platforms, where multiple pharmaceutical companies share infrastructure and applications. The regulated user cannot claim that shared tenancy dilutes their responsibility or that other tenants’ activities absolve them of compliance obligations. Each organization must demonstrate control and oversight as if it were the sole user of the system.
7.2 Audit: Risk-Based Assessment That Actually Means Something
Section 7.2 transforms supplier auditing from an optional risk management exercise into a structured compliance requirement: “When a regulated user is relying on a vendor’s or a service provider’s qualification and/or operation of a system used in GMP activities, the regulated user should, according to risk and system criticality, conduct an audit or a thorough assessment to determine the adequacy of the vendor or service provider’s implemented procedures, the documentation associated with the deliverables, and the potential to leverage these rather than repeating the activities.”
The language “according to risk and system criticality” establishes a scalable framework that requires organizations to classify their systems and adjust audit rigor accordingly. A cloud-based LIMS managing batch release testing demands different scrutiny than a SaaS platform used for training record management. However, the draft makes clear that risk-based does not mean risk-free—even lower-risk systems require documented assessment to justify reduced audit intensity.
The phrase “thorough assessment” provides flexibility for organizations that cannot conduct traditional on-site audits of major cloud providers like AWS or Microsoft. However, it establishes a burden of proof requiring organizations to demonstrate that their assessment methodology provides equivalent assurance to traditional auditing approaches. This might include reviewing third-party certifications, analyzing security documentation, or conducting remote assessments of provider capabilities.
The requirement to evaluate “potential to leverage” supplier documentation acknowledges the reality that many cloud providers and SaaS vendors have invested heavily in GxP-compliant infrastructure and documentation. Organizations can potentially reduce their validation burden by demonstrating that supplier qualifications meet regulatory requirements, but they must affirmatively prove this rather than simply assuming it.
For organizations managing dozens or hundreds of supplier relationships, the audit requirement creates significant resource implications. Companies must develop risk classification methodologies, train audit teams on digital infrastructure assessment, and establish ongoing audit cycles that account for the dynamic nature of cloud services and SaaS platforms.
7.3 Oversight: SLAs and KPIs That Actually Matter
The oversight requirement in Section 7.3 mandates active, continuous supplier management rather than passive relationship maintenance: “When a regulated user is relying on a service provider’s or an internal IT department’s operation of a system used in GMP activities, the regulated user should exercise effective oversight of this according to defined service level agreements (SLA) and key performance indicators (KPI) agreed with the service provider or the internal IT department.”
This requirement acknowledges that traditional supplier management approaches, based on annual reviews and incident-driven interactions, are inadequate for managing dynamic digital services. Cloud platforms undergo continuous updates. SaaS providers deploy new features regularly. Infrastructure changes occur without direct customer notification. The oversight requirement establishes expectations for real-time monitoring and proactive management of these relationships.
The emphasis on “defined” SLAs and KPIs means organizations cannot rely on generic service level commitments provided by suppliers. Instead, they must negotiate specific metrics aligned with GMP requirements and data integrity objectives. For a cloud-based manufacturing execution system, relevant KPIs might include system availability during manufacturing campaigns, data backup completion rates, and incident response times for GMP-critical issues.
Effective oversight requires organizations to establish monitoring systems capable of tracking supplier performance against agreed metrics. This might involve automated dashboard monitoring of system availability, regular review of supplier-provided performance reports, or integration of supplier metrics into internal quality management systems. The goal is continuous visibility into supplier performance rather than retrospective assessment during periodic reviews.
The requirement also applies to internal IT departments, recognizing that many pharmaceutical organizations struggle with accountability when GMP systems are managed by IT teams that don’t report to quality functions. The draft requires the same SLA and KPI framework for internal providers, establishing clear performance expectations and accountability mechanisms.
Evaluating KPIs for IT Service Providers
When building a system of Key Performance Indicators (KPIs) for supplier and service management in a GxP-regulated environment you will want KPIs that truly measure your suppliers’ performance and your own ability to maintain control and regulatory compliance. Since the new requirements emphasize continuous oversight, risk-based evaluation, and lifecycle management, KPIs should cover not just commercial performance but all areas of GxP relevance.
Here are supplier KPIs that are practical, defensible, and ready to justify in both quality forums and to auditors:
1. System Availability/Uptime Measures the percentage of time your supplier’s system or service is fully operational during agreed business hours (or 24/7 for critical GMP systems). Target: 99.9% uptime for critical systems.
2. Incident Response Time Average or maximum time elapsed between a reported incident (especially those affecting GMP/data integrity) and initial supplier response. Target: Immediate acknowledgment; <4 hours for GMP-impacting incidents.
3. Incident Resolution/Recovery Time Average time taken to fully resolve GMP-critical incidents and restore compliant operations. Target: <24 hours for resolution, with root cause and preventive action documented.
4. Change Notification Timeliness Measures whether the supplier notifies you of planned changes, updates, or upgrades within the contractually required timeframe before implementation. Target: 100% advance notification as per contract (e.g., 30 days for non-critical, 48 hours for critical updates).
5. Data Backup Success Rate Percentage of scheduled backups completed successfully and verified for integrity. Target: 100% for GMP-relevant data.
6. Corrective and Preventive Action (CAPA) Closure Rate Percentage of supplier-driven CAPA actions (arising from audits, incidents, or performance monitoring) closed on time. Target: 95% closed within agreed timelines.
7. Audit Finding Closure Timeliness Measures time from audit finding notification to completed remediation (agreed corrective action implemented and verified). Target: 100% of critical findings closed within set period (e.g., 30 days).
8. Percentage of Deliverables On-Time For services involving defined deliverables (e.g., validation documentation, periodic reports)—what percentage arrive within agreed deadlines. Target: 98–100%.
9. Compliance with Change Control Rate at which supplier’s changes (software, hardware, infrastructure) are processed in accordance with your approved change control system—including proper notification, documentation, and assessment. Target: 100% compliance.
10. Regulatory/SLA Audit Support Satisfaction Measured by feedback (internal or from inspectors) on supplier’s effectiveness and readiness in supporting regulatory or SLA-related audits. Target: 100% “satisfactory.”
11. Security Event/Incident Rate Number of security events or potential data integrity breaches attributable to the supplier per reporting period. Target: Zero for GMP-impacting events; rapid supplier notification if any occur.
12. Service Request Resolution Rate Percentage of service/support requests (tickets) resolved within the defined response and resolution SLAs. Target: 98%+.
13. Documentation Accessibility Rate Percentage of required documentation (validation packages, SOPs, certifications, audit trails) available on demand (especially during inspection readiness checks). Target: 100%.
14. Training Completion Rate for Supplier Personnel Percentage of supplier team members assigned to your contract who have successfully completed required GxP and data integrity training. Target: 100%.
To be Annex 11 ready, always align your KPIs with your supplier’s contract (including SLAs/KPIs written into the agreement). Track these metrics and trend them over time—continual improvement and transparency are expected.
Also, regularly review and risk-assess your chosen KPIs: as the risk profile of the supplier or service changes, update the KPIs and targets, and ensure they are embedded into your supplier oversight, quality management review, and audit processes. This forms a defensible part of your data integrity and supplier management evidence under the upcoming draft Annex 11.
7.4 Documentation Availability: No More “Black Box” Services
Section 7.4 addresses one of the most persistent challenges in modern supplier management—ensuring access to documentation needed for regulatory compliance: “When a regulated user relies on a vendor’s, a service provider’s or an internal IT department’s qualification and/or operation of a system used in GMP activities, the regulated user should ensure that documentation for activities required in this document is accessible and can be explained from their facility.”
The phrase “accessible and can be explained” establishes two distinct requirements. Documentation must be physically or electronically available when needed, but organizations must also maintain sufficient understanding to explain systems and processes to regulatory inspectors. This eliminates the common practice of simply collecting supplier documentation without ensuring internal teams understand its contents and implications.
For cloud-based systems, this requirement creates particular challenges. Major cloud providers like AWS, Microsoft Azure, and Google Cloud offer extensive documentation about their infrastructure and services, but pharmaceutical companies must identify which documents are relevant to their specific GMP applications and ensure they can explain how cloud architecture supports data integrity and system reliability.
SaaS providers typically provide less detailed technical documentation, focusing instead on user guides and administrative procedures. Organizations must work with suppliers to obtain validation documentation, system architecture information, and technical specifications needed to demonstrate compliance. This often requires negotiating specific documentation requirements into service agreements rather than accepting standard documentation packages.
The requirement that documentation be explainable “from their facility” means organizations cannot simply reference supplier documentation during inspections. Internal teams must understand system architecture, data flows, security controls, and validation approaches well enough to explain them without direct supplier support. This necessitates significant knowledge transfer from suppliers and ongoing training for internal personnel.
7.5 Contracts: From Legal Formalities to GMP Control Documents
The final subsection transforms supplier contracts from legal formalities into operational control documents: “When a regulated user is relying on a service provider’s or an internal IT department’s qualification and/or operation of a system used in GMP activities, the regulated user should have a contract with a service provider or have approved procedures with an internal IT department which: i. Describes the activities and documentation to be provided ii. Establishes the company procedures and regulatory requirements to be met iii. Agrees on regular, ad hoc and incident reporting and oversight (incl. SLAs and KPIs), answer times, resolution times, etc. iv. Agrees on conditions for supplier audits v. Agrees on support during regulatory inspections, if so requested”
This contract framework establishes five essential elements that transform supplier agreements from commercial documents into GMP control mechanisms. Each element addresses specific compliance risks that have emerged as pharmaceutical organizations increased their reliance on external providers.
Activities and Documentation (7.5.i): This requirement ensures contracts specify exactly what work will be performed and what documentation will be provided. Generic service descriptions become inadequate when regulatory compliance depends on specific activities being performed to defined standards. For a cloud infrastructure provider, this might specify data backup procedures, security monitoring activities, and incident response protocols. For a SaaS platform, it might detail user access management, audit trail generation, and data export capabilities.
Regulatory Requirements (7.5.ii): Contracts must explicitly establish which regulatory requirements apply to supplier activities and how compliance will be demonstrated. This eliminates ambiguity about whether suppliers must meet GxP standards and establishes accountability for regulatory compliance. Suppliers cannot claim ignorance of pharmaceutical requirements, and regulated companies cannot assume suppliers understand applicable standards without explicit contractual clarification.
Reporting and Oversight (7.5.iii): The requirement for “regular, ad hoc and incident reporting” establishes expectations for ongoing communication beyond standard commercial reporting. Suppliers must provide performance data, incident notifications, and ad hoc reports needed for effective oversight. The specification of “answer times” and “resolution times” ensures suppliers commit to response standards aligned with GMP operational requirements rather than generic commercial service levels.
Audit Conditions (7.5.iv): Contracts must establish explicit audit rights and conditions, eliminating supplier claims that audit activities exceed contractual scope. This is particularly important for cloud providers and SaaS vendors who serve multiple industries and may resist pharmaceutical-specific audit requirements. The contractual audit framework must specify frequency, scope, access rights, and supplier support obligations.
Regulatory Inspection Support (7.5.v): Perhaps the most critical requirement, contracts must establish supplier obligations to support regulatory inspections “if so requested.” This cannot be optional or subject to additional fees—it must be a contractual obligation. Suppliers must commit to providing documentation, expert testimony, and system demonstrations needed during regulatory inspections. For cloud providers, this might include architectural diagrams and security certifications. For SaaS vendors, it might include system demonstrations and user access reports.
The Cloud Provider Challenge: Managing Hyperscale Relationships
Section 7’s requirements create particular challenges for organizations using hyperscale cloud providers like Amazon Web Services, Microsoft Azure, and Google Cloud Platform. These providers serve thousands of customers across multiple industries and typically resist customization of their standard service agreements and operational procedures. However, the draft Annex 11 requirements apply regardless of provider size or market position.
Shared Responsibility Models: Cloud providers operate on shared responsibility models where customers retain responsibility for data, applications, and user access while providers manage infrastructure, physical security, and basic services. Section 7 requires pharmaceutical companies to understand and document these responsibility boundaries clearly, ensuring no compliance gaps exist between customer and provider responsibilities.
Standardized Documentation: Hyperscale providers offer extensive documentation about their services, security controls, and compliance certifications. However, pharmaceutical companies must identify which documents are relevant to their specific GMP applications and ensure they understand how provider capabilities support their compliance obligations. This often requires significant analysis of provider documentation to extract GMP-relevant information.
Audit Rights: Traditional audit rights are generally not available with hyperscale cloud providers, who instead offer third-party certifications and compliance reports. Organizations must develop alternative assessment methodologies that satisfy Section 7.2 requirements while acknowledging the realities of cloud provider business models. This might include relying on SOC 2 Type II reports, ISO 27001 certifications, and specialized GxP assessments provided by the cloud provider.
Service Level Agreements: Cloud providers offer standard SLAs focused on technical performance metrics like availability and response times. Pharmaceutical companies must ensure these standard metrics align with GMP requirements or negotiate additional commitments. For example, standard 99.9% availability commitments may be inadequate for systems supporting continuous manufacturing operations.
Incident Response: Cloud provider incident response procedures focus on technical service restoration rather than GMP impact assessment. Organizations must establish internal procedures to evaluate the GMP implications of cloud incidents and ensure appropriate notifications and investigations occur even when the underlying technical issues are resolved by the provider.
SaaS Platform Management: Beyond Standard IT Procurement
Software-as-a-Service platforms present unique challenges under Section 7 because they combine infrastructure management with application functionality, often operated by providers with limited pharmaceutical industry experience. Unlike hyperscale cloud providers who focus purely on infrastructure, SaaS vendors make decisions about application design, user interface, and business workflows that directly impact GMP compliance.
Validation Dependencies: SaaS platforms undergo continuous development and deployment cycles that can affect GMP functionality without customer involvement. Section 7 requires organizations to maintain oversight of these changes and ensure ongoing validation despite dynamic platform evolution. This necessitates change control procedures that account for supplier-initiated modifications and validation strategies that accommodate continuous deployment models.
Data Integrity Controls: SaaS platforms must implement audit trail capabilities, user access controls, and data integrity measures aligned with ALCOA+ principles. However, many platforms designed for general business use lack pharmaceutical-specific features. Organizations must work with suppliers to ensure platform capabilities support GMP requirements or implement compensating controls to address gaps.
Multi-Tenant Considerations: Most SaaS platforms operate in multi-tenant environments where multiple customers share application instances and infrastructure. This creates unique challenges for demonstrating data segregation, ensuring audit trail integrity, and maintaining security controls. Organizations must understand multi-tenant architecture and verify that other tenants cannot access or affect their GMP data.
Integration Management: SaaS platforms typically integrate with other systems through APIs and data feeds that may not be under direct pharmaceutical company control. Section 7 oversight requirements extend to these integrations, requiring organizations to understand data flows, validation status, and change control procedures for all connected systems.
Exit Strategies: The draft Annex 11 implications include requirements for data retrieval and system discontinuation procedures. SaaS contracts must specify data export capabilities, retention periods, and migration support to ensure organizations can maintain compliance during platform transitions.
Internal IT Department Transformation
One of the most significant aspects of Section 7 is its explicit inclusion of internal IT departments within the supplier management framework. This acknowledges the reality that many pharmaceutical organizations have created artificial separations between quality functions and IT support, leading to unclear accountability and inadequate oversight of GMP-critical systems.
Procedural Requirements: The draft requires “approved procedures” with internal IT departments that mirror the contractual requirements applied to external suppliers. This means IT departments must operate under documented procedures that specify their GMP responsibilities, performance expectations, and accountability mechanisms.
SLA Framework: Internal IT departments must commit to defined service level agreements and key performance indicators just like external suppliers. This eliminates the informal, best-effort support models that many organizations have relied upon for internal IT services. IT departments must commit to specific response times, availability targets, and resolution procedures for GMP-critical systems.
Audit and Oversight: Quality organizations must implement formal oversight processes for internal IT departments, including regular performance reviews, capability assessments, and compliance evaluations. This may require establishing new organizational relationships and reporting structures to ensure appropriate independence and accountability.
Change Management: Internal IT departments must implement change control procedures that align with GMP requirements rather than general IT practices. This includes impact assessment procedures, testing requirements, and approval processes that account for potential effects on product quality and data integrity.
Documentation Standards: IT departments must maintain documentation to the same standards required of external suppliers, including system architecture documents, validation records, and operational procedures. This often requires significant upgrades to IT documentation practices and knowledge management systems.
Risk-Based Implementation Strategy
Section 7’s risk-based approach requires organizations to develop systematic methodologies for classifying suppliers and systems, determining appropriate oversight levels, and allocating management resources effectively. This represents a significant departure from one-size-fits-all approaches that many organizations have used for supplier management.
System Criticality Assessment: Organizations must classify their computerized systems based on impact to product quality, patient safety, and data integrity. This classification drives the intensity of supplier oversight, audit requirements, and contractual controls. Critical systems like manufacturing execution systems and laboratory information management systems require the highest level of supplier management, while lower-impact systems like general productivity applications may warrant less intensive oversight.
Supplier Risk Profiling: Different types of suppliers present different risk profiles that affect management approaches. Hyperscale cloud providers typically have robust infrastructure and security controls but limited pharmaceutical industry knowledge. Specialized pharmaceutical software vendors understand GxP requirements but may have less mature operational capabilities. Contract research organizations have pharmaceutical expertise but variable quality systems. Organizations must develop supplier-specific management strategies that account for these different risk profiles.
Audit Planning: Risk-based audit planning requires organizations to prioritize audit activities based on system criticality, supplier risk, and business impact. High-risk suppliers supporting critical systems require comprehensive audits, while lower-risk relationships may be managed through document reviews and remote assessments. Organizations must develop audit scheduling that ensures adequate coverage while managing resource constraints.
Performance Monitoring: Risk-based monitoring means different suppliers require different levels of ongoing oversight. Critical suppliers need real-time performance monitoring and frequent review cycles, while lower-risk suppliers may be managed through periodic assessments and exception reporting. Organizations must implement monitoring systems that provide appropriate visibility without creating excessive administrative burden.
Data Ownership and Access Rights
Section 7’s requirements for clear data ownership and access rights address one of the most contentious issues in modern supplier relationships. Many cloud providers and SaaS vendors have terms of service that create ambiguity about data ownership, retention rights, and access capabilities that are incompatible with GMP requirements.
Ownership Clarity: Contracts must explicitly establish that pharmaceutical companies retain full ownership of all GMP data regardless of where it is stored or processed. This includes not only direct manufacturing and quality data but also metadata, audit trails, and system configuration information. Suppliers cannot claim any ownership rights or use licenses that could affect data availability or integrity.
Access Rights: Pharmaceutical companies must maintain unrestricted access to their data for regulatory purposes, internal investigations, and business operations. This includes both standard data access through application interfaces and raw data access for migration or forensic purposes. Suppliers cannot impose restrictions on data access that could interfere with regulatory compliance or business continuity.
Retention Requirements: Contracts must specify data retention periods that align with pharmaceutical industry requirements rather than supplier standard practices. GMP data may need to be retained for decades beyond normal business lifecycles, and suppliers must commit to maintaining data availability throughout these extended periods.
Migration Rights: Organizations must retain the right to migrate data from supplier systems without restriction or penalty. This includes both planned migrations during contract transitions and emergency migrations necessitated by supplier business failures or service discontinuations. Suppliers must provide data in standard formats and support migration activities.
Regulatory Access: Suppliers must support regulatory inspector access to data and systems as required by pharmaceutical companies. This cannot be subject to additional fees or require advance notice that could delay regulatory compliance. Suppliers must understand their role in regulatory inspections and commit to providing necessary support.
Change Control and Communication
The dynamic nature of cloud services and SaaS platforms creates unique challenges for change control that Section 7 addresses through requirements for proactive communication and impact assessment. Traditional change control models based on formal change requests and approval cycles are incompatible with continuous deployment models used by many digital service providers.
Change Notification: Suppliers must provide advance notification of changes that could affect GMP compliance or system functionality. This includes not only direct application changes but also infrastructure modifications, security updates, and business process changes. The notification period must be sufficient to allow impact assessment and implementation of any necessary mitigating measures.
Impact Assessment: Pharmaceutical companies must evaluate the GMP implications of supplier changes even when the technical impact appears minimal. A cloud provider’s infrastructure upgrade could affect system performance during critical manufacturing operations. A SaaS platform’s user interface change could impact operator training and qualification requirements. Organizations must develop change evaluation procedures that account for these indirect effects.
Emergency Changes: Suppliers must have procedures for emergency changes that balance urgent technical needs with GMP requirements. Security patches and critical bug fixes cannot wait for formal change approval cycles, but pharmaceutical companies must be notified and given opportunity to assess implications. Emergency change procedures must include retroactive impact assessment and documentation requirements.
Testing and Validation: Changes to supplier systems may require re-testing or revalidation of pharmaceutical company applications and processes. Contracts must specify supplier support for customer testing activities and establish responsibilities for validation of changes. This is particularly challenging for multi-tenant SaaS platforms where changes affect all customers simultaneously.
Rollback Capabilities: Suppliers must maintain capabilities to reverse changes that adversely affect GMP compliance or system functionality. This includes technical rollback capabilities and procedural commitments to restore service levels if changes cause operational problems. Rollback procedures must account for data integrity implications and ensure no GMP data is lost or corrupted during restoration activities.
Incident Management and Response
Section 7’s requirements for incident reporting and response acknowledge that service disruptions, security incidents, and system failures have different implications in GMP environments compared to general business applications. Suppliers must understand these implications and adapt their incident response procedures accordingly.
Incident Classification: Suppliers must classify incidents based on GMP impact rather than purely technical severity. A brief database connectivity issue might be low priority from a technical perspective but could affect batch release decisions and require immediate escalation. Suppliers must understand pharmaceutical business processes well enough to assess GMP implications accurately.
Notification Procedures: Incident notification procedures must account for pharmaceutical industry operational patterns and regulatory requirements. Manufacturing operations may run around the clock, requiring immediate notification for GMP-critical incidents. Regulatory reporting obligations may require incident documentation within specific timeframes that differ from standard business practices.
Investigation Support: Suppliers must support pharmaceutical company investigations of incidents that could affect product quality or data integrity. This includes providing detailed technical information, preserving evidence, and making subject matter experts available for investigation activities. Investigation support cannot be subject to additional fees or require formal legal processes.
Corrective Actions: Incident response must include identification and implementation of corrective actions to prevent recurrence. Suppliers must commit to addressing root causes rather than simply restoring service functionality. Corrective action plans must be documented and tracked to completion with pharmaceutical company oversight.
Regulatory Reporting: Suppliers must understand when incidents may require regulatory reporting and provide information needed to support pharmaceutical company reporting obligations. This includes detailed incident timelines, impact assessments, and corrective action documentation. Suppliers must maintain incident records for periods consistent with pharmaceutical industry retention requirements.
Performance Monitoring and Metrics
The oversight requirements in Section 7 necessitate comprehensive performance monitoring systems that go beyond traditional IT service management to encompass GMP-specific requirements and quality metrics. Organizations must implement monitoring frameworks that provide real-time visibility into supplier performance while demonstrating ongoing compliance with regulatory requirements.
GMP-Relevant Metrics: Performance monitoring must include metrics that reflect GMP impact rather than purely technical performance. System availability during manufacturing campaigns is more important than general uptime statistics. Data backup completion rates are more critical than storage utilization metrics. Response times for GMP-critical incidents require different measurement than general support ticket resolution.
Real-Time Monitoring: The dynamic nature of cloud services requires real-time monitoring capabilities rather than periodic reporting. Organizations must implement dashboard systems that provide immediate visibility into supplier performance and alert capabilities for GMP-critical events. This often requires integration between supplier monitoring systems and internal quality management platforms.
Trend Analysis: Performance monitoring must include trend analysis capabilities to identify degrading performance before it affects GMP operations. Gradual increases in system response times could indicate capacity constraints that might affect manufacturing efficiency. Increasing incident frequencies could suggest infrastructure problems that require proactive intervention.
Compliance Metrics: Monitoring systems must track compliance-related metrics such as audit trail completeness, user access control effectiveness, and change control adherence. These metrics require deeper integration with supplier systems and may not be available through standard monitoring interfaces. Organizations may need to negotiate specific compliance reporting capabilities into their service agreements.
Exception Reporting: Performance monitoring must include exception reporting capabilities that identify situations requiring management attention. Missed SLA targets, compliance deviations, and unusual system behavior must trigger immediate notifications and investigation procedures. Exception reporting thresholds must account for GMP operational requirements rather than general business practices.
Audit Trail and Documentation Integration
Section 7’s documentation requirements extend beyond static documents to encompass dynamic audit trail information and real-time system monitoring data that must be integrated with internal quality management systems. This creates significant technical and procedural challenges for organizations managing multiple supplier relationships.
Audit Trail Aggregation: Organizations using multiple suppliers must aggregate audit trail information from various sources to maintain complete records of GMP activities. A manufacturing batch might involve data from cloud-based LIMS systems, SaaS quality management platforms, and locally managed manufacturing execution systems. All audit trail information must be correlated and preserved to support regulatory requirements.
Data Format Standardization: Different suppliers provide audit trail information in different formats and structures, making aggregation and analysis challenging. Organizations must work with suppliers to establish standardized data formats or implement translation capabilities to ensure audit trail information can be effectively integrated and analyzed.
Retention Coordination: Audit trail retention requirements may exceed supplier standard practices, requiring coordination to ensure information remains available throughout required retention periods. Organizations must verify that supplier retention policies align with GMP requirements and establish procedures for retrieving historical audit trail data when needed.
Search and Retrieval: Integrated audit trail systems must provide search and retrieval capabilities that span multiple supplier systems. Regulatory investigations may require analysis of activities across multiple platforms and timeframes. Organizations must implement search capabilities that can effectively query distributed audit trail information.
Access Control Integration: Audit trail access must be controlled through integrated access management systems that span multiple suppliers. Users should not require separate authentication for each supplier system, but access controls must maintain appropriate segregation and monitoring capabilities. This often requires federated identity management systems and single sign-on capabilities.
Validation Strategies for Supplier-Managed Systems
Section 7’s responsibility requirements mean that pharmaceutical companies cannot rely solely on supplier validation activities but must implement validation strategies that encompass supplier-managed systems while avoiding duplication of effort. This requires sophisticated approaches that leverage supplier capabilities while maintaining regulatory accountability.
Hybrid Validation Models: Organizations must develop validation approaches that combine supplier-provided validation evidence with customer-specific testing and verification activities. Suppliers may provide infrastructure qualification documentation, but customers must verify that applications perform correctly on that infrastructure. SaaS providers may offer functional testing evidence, but customers must verify that functionality meets their specific GMP requirements.
Continuous Validation: The dynamic nature of supplier-managed systems requires continuous validation approaches rather than periodic revalidation cycles. Automated testing systems must verify that system functionality remains intact after supplier changes. Monitoring systems must detect performance degradation that could affect validation status. Change control procedures must include validation impact assessment for all supplier modifications.
Risk-Based Testing: Validation testing must focus on GMP-critical functionality rather than comprehensive system testing. Organizations must identify the specific functions that affect product quality and data integrity and concentrate validation efforts on these areas. This requires detailed understanding of business processes and system functionality to determine appropriate testing scope.
Supplier Validation Leverage: Organizations should leverage supplier validation activities where possible while maintaining ultimate responsibility for validation adequacy. This requires assessment of supplier validation procedures, review of testing evidence, and verification that supplier validation scope covers customer GMP requirements. Supplier validation documentation becomes input to customer validation activities rather than replacement for them.
Documentation Integration: Validation documentation must integrate supplier-provided evidence with customer-generated testing results and assessments. The final validation package must demonstrate comprehensive coverage of GMP requirements while clearly delineating supplier and customer contributions to validation activities.
Effective implementation of Section 7 requirements necessitates significant organizational changes that extend beyond traditional supplier management functions to encompass quality assurance, information technology, regulatory affairs, and legal departments. Organizations must develop cross-functional capabilities and governance structures that can manage complex supplier relationships while maintaining regulatory compliance.
Organizational Structure: Many pharmaceutical companies will need to establish dedicated supplier management functions with specific responsibility for GMP-critical supplier relationships. These functions must combine procurement expertise with quality assurance knowledge and technical understanding of computerized systems. Traditional procurement organizations typically lack the regulatory knowledge needed to manage GMP suppliers effectively.
Cross-Functional Teams: Supplier management requires coordination between multiple organizational functions including quality assurance, information technology, regulatory affairs, legal, and procurement. Cross-functional teams must be established to manage complex supplier relationships and ensure all relevant perspectives are considered in supplier selection, contract negotiation, and ongoing oversight activities.
Competency Development: Organizations must develop internal competencies in areas such as cloud infrastructure assessment, SaaS platform evaluation, and contract negotiation for digital services. Many pharmaceutical companies have limited experience in these areas and will need to invest in training and potentially external expertise to build necessary capabilities.
Technology Infrastructure: Effective supplier oversight requires significant technology infrastructure including monitoring systems, audit trail aggregation platforms, and integration capabilities. Organizations must invest in systems that can provide real-time visibility into supplier performance and integrate supplier-provided information with internal quality management systems.
Process Standardization: Supplier management processes must be standardized across the organization to ensure consistent approaches and facilitate knowledge sharing. This includes risk assessment methodologies, audit procedures, contract templates, and performance monitoring frameworks. Standardization becomes particularly important as organizations manage increasing numbers of supplier relationships.
Regulatory Implications and Inspection Readiness
Section 7 requirements significantly change regulatory inspection dynamics by extending inspector access and scrutiny to supplier systems and processes. Organizations must prepare for inspections that encompass their entire supply chain rather than just internal operations, while ensuring suppliers understand and support regulatory compliance obligations.
Extended Inspection Scope: Regulatory inspectors may request access to supplier systems, documentation, and personnel as part of pharmaceutical company inspections. This extends inspection scope beyond traditional facility boundaries to encompass cloud data centers, SaaS platform operations, and supplier quality management systems. Organizations must ensure suppliers understand these obligations and commit to providing necessary support.
Supplier Participation: Suppliers may be required to participate directly in regulatory inspections through system demonstrations, expert testimony, or document provision. This represents a significant change from traditional inspection models where suppliers remained in the background. Suppliers must understand regulatory expectations and prepare to engage directly with inspectors when required.
Documentation Coordination: Inspection preparation must coordinate documentation from multiple suppliers and ensure consistent presentation of integrated systems and processes. This requires significant advance planning and coordination with suppliers to ensure required documentation is available and personnel can explain supplier-managed systems effectively.
Response Coordination: Inspection responses and corrective actions may require coordination with multiple suppliers, particularly when findings relate to integrated systems or shared responsibilities. Organizations must establish procedures for coordinating supplier responses and ensuring corrective actions address root causes across the entire supply chain.
Ongoing Readiness: Inspection readiness becomes a continuous requirement rather than periodic preparation as supplier-managed systems undergo constant change. Organizations must maintain ongoing documentation updates, supplier coordination, and internal knowledge to ensure they can explain and defend their supplier management practices at any time.
Implementation Roadmap and Timeline
Organizations implementing Section 7 requirements must develop comprehensive implementation roadmaps that account for the complexity of modern supplier relationships and the time required to establish new capabilities and procedures. Implementation planning must balance regulatory compliance timelines with practical constraints of supplier negotiation and system modification.
Assessment Phase (Months 1-6): Organizations must begin with comprehensive assessment of current supplier relationships, system dependencies, and gap identification. This includes inventory of all suppliers supporting GMP activities, risk classification of supplier relationships, and evaluation of current contracts and procedures against Section 7 requirements. Assessment activities should identify high-priority gaps requiring immediate attention and longer-term improvements needed for full compliance.
Supplier Engagement (Months 3-12): Parallel to internal assessment, organizations must engage suppliers to communicate new requirements and negotiate contract modifications. This process varies significantly based on supplier type and relationship maturity. Hyperscale cloud providers typically resist contract modifications but may offer additional compliance documentation or services. Specialized pharmaceutical software vendors may be more willing to accommodate specific requirements but may require time to develop new capabilities.
Contract Renegotiation (Months 6-18): Contract modifications to incorporate Section 7 requirements represent major undertakings that may require extensive negotiation and legal review. Organizations should prioritize critical suppliers and high-risk relationships while developing template approaches that can be applied more broadly. Contract renegotiation timelines must account for supplier response times and potential resistance to pharmaceutical-specific requirements.
Procedure Development (Months 6-12): New procedures must be developed for supplier oversight, performance monitoring, audit planning, and incident response. These procedures must integrate with existing quality management systems while accommodating the unique characteristics of different supplier types. Procedure development should include training materials and competency assessment approaches to ensure effective implementation.
Technology Implementation (Months 9-24): Monitoring systems, audit trail aggregation platforms, and integration capabilities require significant technology implementation efforts. Organizations should plan for extended implementation timelines and potential integration challenges with supplier systems. Technology implementation should be phased to address critical suppliers first while building capabilities for broader deployment.
Training and Competency (Months 12-18): Personnel across multiple functions require training on new supplier management approaches and specific competencies for managing different types of supplier relationships. Training programs must be developed for various roles including supplier managers, quality assurance personnel, auditors, and technical specialists. Competency assessment and ongoing training requirements must be established to maintain capabilities as supplier relationships evolve.
Ongoing Monitoring (Continuous): Full implementation of Section 7 requirements establishes ongoing monitoring and continuous improvement processes that become permanent organizational capabilities. Performance monitoring, supplier relationship management, and compliance assessment become routine activities that require sustained resource allocation and management attention.
Future Implications and Industry Evolution
Section 7 represents more than regulatory compliance requirements—it establishes a framework for pharmaceutical industry evolution toward fully integrated digital supply chains where traditional boundaries between internal and external operations become increasingly meaningless. Organizations that successfully implement these requirements will gain competitive advantages through enhanced operational flexibility and risk management capabilities.
Supply Chain Integration: Section 7 requirements drive deeper integration between pharmaceutical companies and their suppliers, creating opportunities for improved efficiency and innovation. Real-time performance monitoring enables proactive management of supply chain risks. Integrated documentation and audit trail systems provide comprehensive visibility into end-to-end processes. Enhanced communication and change management procedures facilitate faster implementation of improvements and innovations.
Technology Evolution: Regulatory requirements for supplier oversight will drive technology innovation in areas such as automated monitoring systems, audit trail aggregation platforms, and integrated validation frameworks. Suppliers will develop pharmaceutical-specific capabilities to meet customer requirements and differentiate their offerings. Technology vendors will emerge to provide specialized solutions for managing complex supplier relationships in regulated industries.
Industry Standards: Section 7 requirements will likely drive development of industry standards for supplier management, contract templates, and integration approaches. Trade associations and standards organizations will develop best practice guidance and template documents to support implementation. Convergence around common approaches will reduce implementation costs and improve interoperability between suppliers and customers.
Regulatory Harmonization: The risk-based, lifecycle-oriented approach embodied in Section 7 aligns with regulatory trends in other jurisdictions and may drive harmonization of global supplier management requirements. FDA Computer Software Assurance guidance shares similar risk-based philosophies, and other regulatory authorities are likely to adopt comparable approaches. Harmonization reduces compliance burden for global pharmaceutical companies and suppliers serving multiple markets.
Competitive Differentiation: Organizations that excel at supplier management under Section 7 requirements will gain competitive advantages through reduced risk, improved operational efficiency, and enhanced innovation capabilities. Effective supplier partnerships enable faster implementation of new technologies and more agile responses to market opportunities. Strong supplier relationships provide resilience during disruptions and enable rapid scaling of operations.
Conclusion: The Strategic Imperative
Section 7 of the draft Annex 11 represents the most significant change in pharmaceutical supplier management requirements since the introduction of 21CFRPart11. The transformation from perfunctory oversight to comprehensive management reflects the reality that modern pharmaceutical operations depend fundamentally on external providers for capabilities that directly affect product quality and patient safety.
Organizations that approach Section 7 implementation as mere regulatory compliance will miss the strategic opportunity these requirements represent. The enhanced supplier management capabilities required by Section 7 enable pharmaceutical companies to leverage external innovation more effectively, manage operational risks more comprehensively, and respond to market opportunities more rapidly than traditional approaches allow.
However, successful implementation requires sustained commitment and significant investment in organizational capabilities, technology infrastructure, and relationship management. Organizations cannot simply modify existing procedures—they must fundamentally reconceptualize their approach to supplier relationships and develop entirely new competencies for managing digital supply chains.
The implementation timeline for Section 7 requirements extends well beyond the expected 2026 effective date for the final Annex 11. Organizations that begin implementation now will have competitive advantages through enhanced capabilities and supplier relationships. Those that delay implementation will find themselves struggling to achieve compliance while their competitors demonstrate regulatory leadership through proactive adoption.
Section 7 acknowledges that pharmaceutical manufacturing has evolved from discrete operations conducted within company facilities to integrated processes that span multiple organizations and geographic locations. Regulatory compliance must evolve correspondingly to encompass these extended operations while maintaining the rigor and accountability that ensures product quality and patient safety.
The future of pharmaceutical manufacturing belongs to organizations that can effectively manage complex supplier relationships while maintaining regulatory compliance and operational excellence. Section 7 provides the framework for this evolution—organizations that embrace it will thrive, while those that resist it will find themselves increasingly disadvantaged in a digitized, interconnected industry.
The message of Section 7 is clear: supplier management is no longer a support function but a core competency that determines organizational success in the modern pharmaceutical industry. Organizations that recognize this reality and invest accordingly will build sustainable competitive advantages that extend far beyond regulatory compliance to encompass operational excellence, innovation capability, and strategic flexibility.
The transformation required by Section 7 is comprehensive and challenging, but it positions the pharmaceutical industry for a future where effective supplier partnerships enable better medicines, safer products, and more efficient operations. Organizations that master these requirements will lead industry evolution toward more innovative, efficient, and patient-focused pharmaceutical development and manufacturing.
Requirement Area
Current Annex 11 (2011)
Draft Annex 11 Section 7 (2025)
Scope of Supplier Management
Third parties (suppliers, service providers) for systems/services
All vendors, service providers, internal IT departments for GMP systems
MAH/Manufacturer Responsibility
Basic – formal agreements must exist
Regulated user remains fully responsible regardless of outsourcing
Risk-Based Assessment
Audit need based on risk assessment
Audit/assessment required according to risk and system criticality
Supplier Qualification Process
Competence and reliability key factors
Detailed qualification with thorough assessment of procedures/documentation
Written Agreements/Contracts
Formal agreements with clear responsibilities
Comprehensive contracts with specific GMP responsibilities defined
Audit Requirements
Risk-based audit decisions
Risk-based audits with defined conditions and support requirements
Ongoing Oversight
Not explicitly detailed
Effective oversight via SLAs and KPIs with defined reporting
Change Management
Not specified
Proactive change notification and assessment requirements
Data Ownership & Access
Not explicitly addressed
Clear data ownership, backup, retention responsibilities in contracts
Documentation Availability
Documentation should be available to inspectors
All required documentation must be accessible and explainable
Service Level Agreements
Not mentioned
Mandatory SLAs with KPIs, reporting, and oversight mechanisms
The strategic utilization of supplier documentation in qualification processes presents a significant opportunity to enhance efficiency while maintaining strict quality standards. Determining what supplier documentation can be accepted and what aspects require additional qualification is critical for streamlining validation activities without compromising product quality or patient safety.
Regulatory Framework Supporting Supplier Documentation Use
Regulatory bodies increasingly recognize the value of leveraging third-party documentation when properly evaluated and integrated into qualification programs. The FDA’s 2011 Process Validation Guidance embraces risk-based approaches that focus resources on critical aspects rather than duplicating standard testing. This guidance references the ASTM E2500 standard, which explicitly addresses the use of supplier documentation in qualification activities.
The EU GMP Annex 15 provides clear regulatory support, stating: “Data supporting qualification and/or validation studies which were obtained from sources outside of the manufacturers own programmes may be used provided that this approach has been justified and that there is adequate assurance that controls were in place throughout the acquisition of such data.” This statement offers a regulatory pathway for incorporating supplier documentation, provided proper controls and justification exist.
ICH Q9 further supports this approach by encouraging risk-based allocation of resources, allowing companies to focus qualification efforts on areas of highest risk while leveraging supplier documentation for well-controlled, lower-risk aspects. The integration of these regulatory perspectives creates a framework that enables efficient qualification strategies while maintaining regulatory compliance.
Benefits of Utilizing Supplier Documentation in Qualification
Biotech manufacturing systems present unique challenges due to their complexity, specialized nature, and biological processes. Leveraging supplier documentation offers multiple advantages in this context:
Supplier expertise in specialized biotech equipment often exceeds that available within pharmaceutical companies. This expertise encompasses deep understanding of complex technologies such as bioreactors, chromatography systems, and filtration platforms that represent years of development and refinement. Manufacturers of bioprocess equipment typically employ specialists who design and test equipment under controlled conditions unavailable to end users.
Integration of engineering documentation into qualification protocols can reduce project timelines, while significantly decreasing costs associated with redundant testing. This efficiency is particularly valuable in biotech, where manufacturing systems frequently incorporate numerous integrated components from different suppliers.
By focusing qualification resources on truly critical aspects rather than duplicating standard supplier testing, organizations can direct expertise toward product-specific challenges and integration issues unique to their manufacturing environment. This enables deeper verification of critical aspects that directly impact product quality rather than dispersing resources across standard equipment functionality tests.
Criteria for Acceptable Supplier Documentation
Audit of the Supplier
Supplier Quality System Assessment
Before accepting any supplier documentation, a thorough assessment of the supplier’s quality system must be conducted. This assessment should evaluate the following specific elements:
Quality management systems certification to relevant standards with verification of certification scope and validity. This should include review of recent certification audit reports and any major findings.
Document control systems that demonstrate proper version control, appropriate approvals, secure storage, and systematic review and update cycles. Specific attention should be paid to engineering document management systems and change control procedures for technical documentation.
Training programs with documented evidence of personnel qualification, including training matrices showing alignment between job functions and required training. Training records should demonstrate both initial training and periodic refresher training, particularly for personnel involved in critical testing activities.
Change control processes with formal impact assessments, appropriate review levels, and implementation verification. These processes should specifically address how changes to equipment design, software, or testing protocols are managed and documented.
Deviation management systems with documented root cause analysis, corrective and preventive actions, and effectiveness verification. The system should demonstrate formal investigation of testing anomalies and resolution of identified issues prior to completion of supplier testing.
Test equipment calibration and maintenance programs with NIST-traceable standards, appropriate calibration frequencies, and out-of-tolerance investigations. Records should demonstrate that all test equipment used in generating qualification data was properly calibrated at the time of testing.
Software validation practices aligned with GAMP5 principles, including risk-based validation approaches for any computer systems used in equipment testing or data management. This should include validation documentation for any automated test equipment or data acquisition systems.
Internal audit processes with independent auditors, documented findings, and demonstrable follow-up actions. Evidence should exist that the supplier conducts regular internal quality audits of departments involved in equipment design, manufacturing, and testing.
Technical Capability Verification
Supplier technical capability must be verified through:
Documentation of relevant experience with similar biotech systems, including a portfolio of comparable projects successfully completed. This should include reference installations at regulated pharmaceutical or biotech companies with complexity similar to the proposed equipment.
Technical expertise of key personnel demonstrated through formal qualifications, industry experience, and specific expertise in biotech applications. Review should include CVs of key personnel who will be involved in equipment design, testing, and documentation.
Testing methodologies that incorporate scientific principles, appropriate statistics, and risk-based approaches. Documentation should demonstrate test method development with sound scientific rationales and appropriate controls.
Calibrated and qualified test equipment with documented measurement uncertainties appropriate for the parameters being measured. This includes verification that measurement capabilities exceed the required precision for critical parameters by an appropriate margin.
GMP understanding demonstrated through documented training, experience in regulated environments, and alignment of test protocols with GMP principles. Personnel should demonstrate awareness of regulatory requirements specific to biotech applications.
Measurement traceability to national standards with documented calibration chains for all critical measurements. This should include identification of reference standards used and their calibration status.
Design control processes aligned with recognized standards including design input review, risk analysis, design verification, and design validation. Design history files should be available for review to verify systematic development approaches.
Documentation Quality Requirements
Acceptable supplier documentation must demonstrate:
Creation under GMP-compliant conditions with evidence of training for personnel generating the documentation. Records should demonstrate that personnel had appropriate training in documentation practices and understood the criticality of accurate data recording.
Compliance with GMP documentation practices including contemporaneous recording, no backdating, proper error correction, and use of permanent records. Documents should be reviewed for evidence of proper data recording practices such as signed and dated entries, proper correction of errors, and absence of unexplained gaps.
Completeness with clearly defined acceptance criteria established prior to testing. Pre-approved protocols should define all test parameters, conditions, and acceptance criteria without post-testing modifications.
Actual test results rather than summary statements, with raw data supporting reported values. Testing documentation should include actual measured values, not just pass/fail determinations, and should provide sufficient detail to allow independent evaluation.
Deviation records with thorough investigations and appropriate resolutions. Any testing anomalies should be documented with formal investigations, root cause analysis, and justification for any retesting or data exclusion.
Traceability to requirements through clear linkage between test procedures and equipment specifications. Each test should reference the specific requirement or specification it is designed to verify.
Authorization by responsible personnel with appropriate signatures and dates. Documents should demonstrate review and approval by qualified individuals with defined responsibilities in the testing process.
Data integrity controls including audit trails for electronic data, validated computer systems, and measures to prevent unauthorized modification. Evidence should exist that data security measures were in place during testing and documentation generation.
Statistical analysis and justification where appropriate, particularly for performance data involving multiple measurements or test runs. Where sampling is used, justification for sample size and statistical power should be provided.
Adherence to established industry standards and design codes relevant to biotech equipment. This includes documentation citing specific standards applied during design and evidence of compliance verification.
Implementation of systematic design methodologies including requirements gathering, conceptual design, detailed design, and design review phases. Design documentation should demonstrate progression through formal design stages with appropriate approvals at each stage.
Application of appropriate testing protocols based on equipment type, criticality, and intended use. Testing strategies should be aligned with industry norms for similar equipment and demonstrate appropriate rigor.
Maintenance of equipment calibration throughout testing phases with records demonstrating calibration status. All test equipment should be documented as calibrated before and after critical testing activities.
Documentation accuracy and completeness demonstrated through systematic review processes and quality checks. Evidence should exist of multiple review levels for critical documentation and formal approval processes.
Implementation of appropriate commissioning procedures aligned with recognized industry practices. Commissioning plans should demonstrate systematic verification of all equipment functions and utilities.
Formal knowledge transfer processes ensuring proper communication between design, manufacturing, and qualification teams. Evidence should exist of structured handover meetings or documentation between project phases.
Types of Supplier Documentation That Can Be Leveraged
When the above criteria are met, the following specific types of supplier documentation can potentially be leveraged.
Factory Acceptance Testing (FAT)
FAT documentation represents comprehensive testing at the supplier’s site before equipment shipment. These documents are particularly valuable because they often represent testing under more controlled conditions than possible at the installation site. For biotech applications, FAT documentation may include:
Functional testing of critical components with detailed test procedures, actual measurements, and predetermined acceptance criteria. This should include verification of all critical operating parameters under various operating conditions.
Control system verification through systematic testing of all control loops, alarms, and safety interlocks. Testing should demonstrate proper response to normal operating conditions as well as fault scenarios.
Material compatibility confirmation with certificates of conformance for product-contact materials and testing to verify absence of leachables or extractables that could impact product quality.
Cleaning system performance verification through spray pattern testing, coverage verification, and drainage evaluation. For CIP (Clean-in-Place) systems, this should include documented evidence of cleaning effectiveness.
Performance verification under load conditions that simulate actual production requirements, with test loads approximating actual product characteristics where possible.
Alarm and safety feature testing with verification of proper operation of all safety interlocks, emergency stops, and containment features critical to product quality and operator safety.
Software functionality testing with documented verification of all user requirements related to automation, control systems, and data management capabilities.
Site Acceptance Testing (SAT)
SAT documentation verifies proper installation and basic functionality at the end-user site. For biotech equipment, this might include:
Installation verification confirming proper utilities connections, structural integrity, and physical alignment according to engineering specifications. This should include verification of spatial requirements and accessibility for operation and maintenance.
Basic functionality testing demonstrating that all primary equipment functions operate as designed after transportation and installation. Tests should verify that no damage occurred during shipping and installation.
Communication with facility systems verification, including integration with building management systems, data historians, and centralized control systems. Testing should confirm proper data transfer and command execution between systems.
Initial calibration verification for all critical instruments and control elements, with documented evidence of calibration accuracy and stability.
Software configuration verification showing proper installation of control software, correct parameter settings, and appropriate security configurations.
Environmental conditions verification confirming that the installed location meets requirements for temperature, humidity, vibration, and other environmental factors that could impact equipment performance.
Design Documentation
Design documents that can support qualification include:
Design specifications with detailed engineering requirements, operating parameters, and performance expectations. These should include rationales for critical design decisions and risk assessments supporting design choices.
Material certificates, particularly for product-contact parts, with full traceability to raw material sources and manufacturing processes. Documentation should include testing for biocompatibility where applicable.
Software design specifications with detailed functional requirements, system architecture, and security controls. These should demonstrate structured development approaches with appropriate verification activities.
Risk analyses performed during design, including FMEA (Failure Mode and Effects Analysis) or similar systematic evaluations of potential failure modes and their impacts on product quality and safety.
Design reviews and approvals with documented participation of subject matter experts across relevant disciplines including engineering, quality, manufacturing, and validation.
Finite element analysis reports or other engineering studies supporting critical design aspects such as pressure boundaries, mixing efficiency, or temperature distribution.
Method Validation and Calibration Documents
For analytical instruments and measurement systems, supplier documentation might include:
Calibration certificates with traceability to national standards, documented measurement uncertainties, and verification of calibration accuracy across the operating range.
Method validation reports demonstrating accuracy, precision, specificity, linearity, and robustness for analytical methods intended for use with the equipment.
Reference standard certifications with documented purity, stability, and traceability to compendial standards where applicable.
Instrument qualification protocols (IQ/OQ) with comprehensive testing of all critical functions and performance parameters against predetermined acceptance criteria.
Software validation documentation showing systematic verification of all calculation algorithms, data processing functions, and reporting capabilities.
What Must Still Be Qualified By The End User
Despite the value of supplier documentation, certain aspects always require direct qualification by the end user. These areas should be the focus of end-user qualification activities:
Facility utility connections and performance verification under actual operating conditions. This must include verification that utilities (water, steam, gases, electricity) meet the required specifications at the point of use, not just at the utility generation source.
Integration with other manufacturing systems, particularly verification of interfaces between equipment from different suppliers. Testing should verify proper data exchange, sequence control, and coordinated operation during normal production and exception scenarios.
Facility-specific environmental conditions including temperature mapping, particulate monitoring, and pressure differentials that could impact biotech processes. Testing should verify that environmental conditions remain within acceptable limits during worst-case operating scenarios.
Network connectivity and data transfer verification, including security controls, backup systems, and disaster recovery capabilities. Testing should demonstrate reliable performance under peak load conditions and proper handling of network interruptions.
Alarm systems integration with central monitoring and response protocols, including verification of proper notification pathways and escalation procedures. Testing should confirm appropriate alarm prioritization and notification of responsible personnel.
Building management system interfaces with verification of environmental monitoring and control capabilities critical to product quality. Testing should verify proper feedback control and response to excursions.
Process-specific parameters beyond standard equipment functionality, with testing under actual operating conditions using representative materials. Testing should verify equipment performance with actual process materials, not just test substances.
Custom configurations for specific products, including verification of specialized equipment settings, program parameters, or mechanical adjustments unique to the user’s products.
Production-scale performance verification, with particular attention to scale-dependent parameters such as mixing efficiency, heat transfer, and mass transfer. Testing should verify that performance characteristics demonstrated at supplier facilities translate to full-scale production.
Process-specific cleaning verification, including worst-case residue removal studies and cleaning cycle development specific to the user’s products. Testing should demonstrate effective cleaning of all product-contact surfaces with actual product residues.
Specific operating ranges for the user’s process, with verification of performance at the extremes of normal operating parameters. Testing should verify capability to maintain critical parameters within required tolerances throughout production cycles.
Process-specific automation sequences and recipes with verification of all production scenarios, including exception handling and recovery procedures. Testing should verify all process recipes and automated sequences with actual production materials.
Hold time verification for intermediate process steps specific to the user’s manufacturing process. Testing should confirm product stability during maximum expected hold times between process steps.
Critical Quality Attributes
Testing related directly to product-specific critical quality attributes should generally not be delegated solely to supplier documentation, particularly for:
Bioburden and endotoxin control verification using the actual production process and materials. Testing should verify absence of microbial contamination and endotoxin introduction throughout the manufacturing process.
Product contact material compatibility studies with the specific products and materials used in production. Testing should verify absence of leachables, extractables, or product degradation due to contact with equipment surfaces.
Product-specific recovery rates and process yields based on actual production experience. Testing should verify consistency of product recovery across multiple batches and operating conditions.
Process-specific impurity profiles with verification that equipment design and operation do not introduce or magnify impurities. Testing should confirm that impurity clearance mechanisms function as expected with actual production materials.
Sterility assurance measures specific to the user’s aseptic processing approaches. Testing should verify the effectiveness of sterilization methods and aseptic techniques with the actual equipment configuration and operating procedures.
Product stability during processing with verification that equipment operation does not negatively impact critical quality attributes. Testing should confirm that product quality parameters remain within acceptable limits throughout the manufacturing process.
Process-specific viral clearance capacity for biological manufacturing processes. Testing should verify effective viral removal or inactivation capabilities with the specific operating parameters used in production.
Operational and Procedural Integration
A critical area often overlooked in qualification plans is operational and procedural integration, which requires end-user qualification for:
Operator interface verification with confirmation that user interactions with equipment controls are intuitive, error-resistant, and aligned with standard operating procedures. Testing should verify that operators can effectively control the equipment under normal and exception conditions.
Procedural workflow integration ensuring that equipment operation aligns with established manufacturing procedures and documentation systems. Testing should verify compatibility between equipment operation and procedural requirements.
Training effectiveness verification for operators, maintenance personnel, and quality oversight staff. Assessment should confirm that personnel can effectively operate, maintain, and monitor equipment in compliance with established procedures.
Maintenance accessibility and procedural verification to ensure that preventive maintenance can be performed effectively without compromising product quality. Testing should verify that maintenance activities can be performed as specified in supplier documentation.
Sampling accessibility and technique verification to ensure representative samples can be obtained safely without compromising product quality. Testing should confirm that sampling points are accessible and provide representative samples.
Change management procedures specific to the user’s quality system, with verification that equipment changes can be properly evaluated, implemented, and documented. Testing should confirm integration with the user’s change control system.
Implementing a Risk-Based Approach to Supplier Documentation
A systematic risk-based approach should be implemented to determine what supplier documentation can be leveraged and what requires additional verification:
Perform impact assessment to categorize system components based on their potential impact on product quality:
Direct impact components with immediate influence on critical quality attributes
Indirect impact components that support direct impact systems
No impact components without reasonable influence on product quality
Conduct risk analysis using formal tools such as FMEA to identify:
Critical components and functions requiring thorough qualification
Identify gaps between supplier documentation and qualification requirements by:
Mapping supplier testing to user requirements
Evaluating the quality and completeness of supplier testing
Identifying areas where supplier testing does not address user-specific requirements
Assessing the reliability and applicability of supplier data to the user’s specific application
Create targeted verification plans to address:
High-risk areas not adequately covered by supplier documentation
User-specific requirements not addressed in supplier testing
Integration points between supplier equipment and user systems
Process-specific performance requirements
This risk-based methodology ensures that qualification resources are focused on areas of highest concern while leveraging reliable supplier documentation for well-controlled aspects.
Documentation and Justification Requirements
When using supplier documentation in qualification, proper documentation and justification are essential:
Create a formal supplier assessment report documenting:
Evaluation methodology and criteria used to assess the supplier
Evidence of supplier quality system effectiveness
Verification of supplier technical capabilities
Assessment of documentation quality and completeness
Identification of any deficiencies and their resolution
Develop a gap assessment identifying:
Areas where supplier documentation meets qualification requirements
Areas requiring additional end-user verification
Rationale for decisions on accepting or supplementing supplier documentation
Risk-based justification for the scope of end-user qualification activities
Prepare a traceability matrix showing:
Mapping between user requirements and testing activities
Source of verification for each requirement (supplier or end-user testing)
Evidence of test completion and acceptance
Cross-references to specific documentation supporting requirement verification
Maintain formal acceptance of supplier documentation with:
Quality unit review and approval of supplier documentation
Documentation of any additional verification activities performed
Records of any deficiencies identified and their resolution
Evidence of conformance to predetermined acceptance criteria
Document rationale for accepting supplier documentation:
Risk-based justification for leveraging supplier testing
Assessment of supplier documentation reliability and completeness
Evaluation of supplier testing conditions and their applicability
Formal incorporation of supplier documentation into the quality system
Version control and change management for supplier documentation
Secure storage and retrieval systems for qualification records
Maintenance of complete documentation packages supporting qualification decisions
Biotech-Specific Considerations
For Cell Culture Systems:
While basic temperature, pressure, and mixing capabilities may be verified through supplier testing, product-specific parameters require end-user verification. These include:
Cell viability and growth characteristics with the specific cell lines used in production. End-user testing should verify consistent cell growth, viability, and productivity under normal operating conditions.
Metabolic profiles and nutrient consumption rates specific to the production process. Testing should confirm that equipment design supports appropriate nutrient delivery and waste removal for optimal cell performance.
Homogeneity studies for bioreactors under process-specific conditions including actual media formulations, cell densities, and production phase operating parameters. Testing should verify uniform conditions throughout the bioreactor volume during all production phases.
Cell culture monitoring systems calibration and performance with actual production cell lines and media. Testing should confirm reliable and accurate monitoring of critical culture parameters throughout the production cycle.
Scale-up effects specific to the user’s cell culture process, with verification that performance characteristics demonstrated at smaller scales translate to production scale. Testing should verify comparable cell growth kinetics and product quality across scales.
For Purification Systems
Chromatography system pressure capabilities and gradient formation may be accepted from supplier testing, but product-specific performance requires end-user verification:
Product-specific recovery, impurity clearance, and yield verification using actual production materials. Testing should confirm consistent product recovery and impurity removal across multiple cycles.
Resin lifetime and performance stability with the specific products and buffer systems used in production. Testing should verify consistent performance throughout the expected resin lifetime.
Cleaning and sanitization effectiveness specific to the user’s products and contaminants. Testing should confirm complete removal of product residues and effective sanitization between production cycles.
Column packing reproducibility and performance with production-scale columns and actual resins. Testing should verify consistent column performance across multiple packing cycles.
Buffer preparation and delivery system performance with actual buffer formulations. Testing should confirm accurate preparation and delivery of all process buffers under production conditions.
For Analytical Methods
Basic instrument functionality can be verified through supplier IQ/OQ documentation, but method-specific performance requires end-user verification:
Method-specific performance with actual product samples, including verification of specificity, accuracy, and precision with the user’s products. Testing should confirm reliable analytical performance with actual production materials.
Method robustness under the specific laboratory conditions where testing will be performed. Testing should verify consistent method performance across the range of expected operating conditions.
Method suitability for the intended use, including capability to detect relevant product variants and impurities. Testing should confirm that the method can reliably distinguish between acceptable and unacceptable product quality.
Operator technique verification to ensure consistent method execution by all analysts who will perform the testing. Assessment should confirm that all analysts can execute the method with acceptable precision and accuracy.
Data processing and reporting verification with the user’s specific laboratory information management systems. Testing should confirm accurate data transfer, calculations, and reporting.
Practical Examples
Example 1: Bioreactor Qualification
For a 2000L bioreactor system, supplier documentation might be leveraged for:
Acceptable with minimal verification: Pressure vessel certification, welding documentation, motor specification verification, basic control system functionality, standard safety features. These aspects are governed by well-established engineering standards and can be reliably verified by the supplier in a controlled environment.
Acceptable with targeted verification: Temperature control system performance, basic mixing capability, sensor calibration procedures. While these aspects can be largely verified by the supplier, targeted verification in the user’s facility ensures that performance meets process-specific requirements.
Requiring end-user qualification: Process-specific mixing studies with actual media, cell culture growth performance, specific gas transfer rates, cleaning validation with product residues. These aspects are highly dependent on the specific process and materials used and cannot be adequately verified by the supplier.
In all cases, the acceptance of supplier documentation must be documented well and performed according to GMPs and at appropriately described in the Validation Plan or other appropriate testing rationale document.
Example 2: Chromatography System Qualification
For a multi-column chromatography system, supplier documentation might be leveraged as follows:
Acceptable with minimal verification: Pressure testing of flow paths, pump performance specifications, UV detector linearity, conductivity sensor calibration, valve switching accuracy. These aspects involve standard equipment functionality that can be reliably verified by the supplier using standardized testing protocols.
Acceptable with targeted verification: Gradient formation accuracy, column switching precision, UV detection sensitivity with representative proteins, system cleaning procedures. These aspects require verification with materials similar to those used in production but can largely be addressed through supplier testing with appropriate controls.
Requiring end-user qualification: Product-specific binding capacity, elution conditions optimization, product recovery rates, impurity clearance, resin lifetime with actual process streams, cleaning validation with actual product residues. These aspects are highly process-specific and require testing with actual production materials under normal operating conditions.
The qualification approach must balance efficiency with appropriate rigor, focusing end-user testing on aspects that are process-specific or critical to product quality.
Example 3: Automated Analytical Testing System Qualification
For an automated high-throughput analytical testing platform used for product release testing, supplier documentation might be leveraged as follows:
Acceptable with minimal verification: Mechanical subsystem functionality, basic software functionality, standard instrument calibration, electrical safety features, standard data backup systems. These fundamental aspects of system performance can be reliably verified by the supplier using standardized testing protocols.
Acceptable with targeted verification: Sample throughput rates, basic method execution, standard curve generation, basic system suitability testing, data export functions. These aspects require verification with representative materials but can largely be addressed through supplier testing with appropriate controls.
Requiring end-user qualification: Method-specific performance with actual product samples, detection of product-specific impurities, method robustness under laboratory-specific conditions, integration with laboratory information management systems, data integrity controls specific to the user’s quality system, analyst training effectiveness. These aspects are highly dependent on the specific analytical methods, products, and laboratory environment.
For analytical systems involved in release testing, additional considerations include:
Verification of method transfer from development to quality control laboratories
Demonstration of consistent performance across multiple analysts
Confirmation of data integrity throughout the complete testing process
Integration with the laboratory’s sample management and result reporting systems
Alignment with regulatory filing commitments for analytical methods
This qualification strategy ensures that standard instrument functionality is efficiently verified through supplier documentation while focusing end-user resources on the product-specific aspects critical to reliable analytical results.
Conclusion: Best Practices for Supplier Documentation in Biotech Qualification
To maximize the benefits of supplier documentation while ensuring regulatory compliance in biotech qualification:
Develop clear supplier requirements early in the procurement process, with specific documentation expectations communicated before equipment design and manufacturing. These requirements should specifically address documentation format, content, and quality standards.
Establish formal supplier assessment processes with clear criteria aligned with regulatory expectations and internal quality standards. These assessments should be performed by multidisciplinary teams including quality, engineering, and manufacturing representatives.
Implement quality agreements with key equipment suppliers, explicitly defining responsibilities for documentation, testing, and qualification activities. These agreements should include specifics on documentation standards, testing protocols, and data integrity requirements.
Create standardized processes for reviewing and accepting supplier documentation based on criticality and risk assessment. These processes should include formal gap analysis and identification of supplemental testing requirements.
Apply risk-based approaches consistently when determining what can be leveraged, focusing qualification resources on aspects with highest potential impact on product quality. Risk assessments should be documented with clear rationales for acceptance decisions.
Document rationale thoroughly for acceptance decisions, including scientific justification and regulatory considerations. Documentation should demonstrate a systematic evaluation process with appropriate quality oversight.
Maintain appropriate quality oversight throughout the process, with quality unit involvement in key decisions regarding supplier documentation acceptance. Quality representatives should review and approve supplier assessment reports and qualification plans.
Implement verification activities targeting gaps and high-risk areas identified during document review, focusing on process-specific and integration aspects. Verification testing should be designed to complement, not duplicate, supplier testing.
Integrate supplier documentation within your qualification lifecycle approach, establishing clear linkages between supplier testing and overall qualification requirements. Traceability matrices should demonstrate how supplier documentation contributes to meeting qualification requirements.
The key is finding the right balance between leveraging supplier expertise and maintaining appropriate end-user verification of critical aspects that impact product quality and patient safety. Proper evaluation and integration of supplier documentation represents a significant opportunity to enhance qualification efficiency while maintaining the rigorous standards essential for biotech products. With clear criteria for acceptance, systematic risk assessment, and thorough documentation, organizations can confidently leverage supplier documentation as part of a comprehensive qualification strategy aligned with current regulatory expectations and quality best practices.
Single-use systems (SUS) have become increasingly prevalent in biopharmaceutical manufacturing due to their flexibility, reduced contamination risk, and cost-effectiveness. The thing is, management of the life-cycle of single-use systems becomes critical and is an area organizations can truly screw up by cutting corners. To do it right requires careful collaboration between all stakeholders in the supply chain, from raw material suppliers to end users.
Design and Development
Apply Quality by Design (QbD) principles from the outset by focusing on process understanding and the design space to create controlled and consistent manufacturing processes that result in high-quality, efficacious products. This approach should be applied to SUS design.
ASTM E3051 “Standard guide for specification, design, verification, and application of SUS in pharmaceutical and biopharmaceutical manufacturing” provides an excellent framework for the design process.
Make sure to conduct thorough risk assessments, considering potential failure modes and effects throughout the SUS life-cycle.
Engage end-users early to understand their specific requirements and process constraints. A real mistake in organizations is not involving the end-users early enough. From the molecule steward to manufacturing these users are critical.
Raw Material and Component Selection
Carefully evaluate and qualify raw materials and components. Work closely with suppliers to understand material properties, extractables/leachables profiles, and manufacturing processes.
Develop comprehensive specifications for critical materials and components. ASTM E3244 is handy place to look for guidance on raw material qualification for SUS.
Manage the Supplier through Manufacturing and Assembly
Implementing robust supplier qualification and auditing programs and establish change control agreements with suppliers to be notified of any changes that could impact SUS performance or quality. It is important the supplier have a robust quality management system and that they apply Good Manufacturing Practices (GMP) through their facilities. Ensure they have in place appropriate controls to
Validate sterilization processes
Conduct routine bioburden and endotoxin testing
Design packaging to protect SUS during transportation and storage. Shipping methods need to protect against physical damage and temperature excursions
Establish appropriate storage conditions and shelf-life based on stability studies
Provide appropriate labeling and traceability
Have appropriate inventory controls. Ideally select suppliers who understand the importance of working with you for collaborative planning, forecasting and replenishment (CPFR)
Testing and Qualification
Develop a comprehensive testing strategy, including integrity testing and conduct extractables and leachables studies following industry guidelines. Evaluate the suppliers shipping and transportation studies to evaluate SUS robustness and determine if you need additional studies.
Implementation and Use
End users should have appropriate and comprehensive documentation and training to end users on proper handling, installation, and use of SUS. These procedures should include how to perform pre-use integrity testing at the point of use as well as how to perform thorough in-process and final inspections.
Consider implementing automated visual inspection systems and other appropriate monitoring.
Implement appropriate environmental monitoring programs in SUS manufacturing areas. While the dream of manufacturing outdoors is a good one, chances are we aren’t even close yet. Don’t short this layer of control.
Continuous Improvement
Ensure you have appropriate mechanisms in place to gather data on SUS performance and any issues encountered during use. Share relevant information across the supply chain to drive improvements.
Conduct periodic audits of suppliers and manufacturing facilities.
Stay updated on evolving regulatory guidance and industry best practices. There is still a lot changing in this space.