The recent FDA warning letter to Sanofi highlights a critical issue in biopharmaceutical manufacturing: the integrity of single-use systems (SUS) and the prevention of leaks. This incident serves as a stark reminder of the importance of robust control strategies in bioprocessing, particularly when it comes to high-pressure events and product leakage.
The Sanofi Case: A Cautionary Tale
In January 2025, the FDA issued a warning letter to Sanofi regarding their Genzyme facility in Framingham, Massachusetts. The letter cited significant deviations from Current Good Manufacturing Practice (CGMP) for active pharmaceutical ingredients (APIs). One of the key issues highlighted was the company’s failure to address high-pressure events that resulted in in-process product leakage.
Sanofi had been using an unapproved workaround, replacing shipping bags to control the frequency of high-pressure and in-process leaking events. This deviation was not properly documented or the solution validated.
A proper control strategy in this context would likely involve:
A validated process modification to prevent or mitigate high-pressure events
Engineering controls or equipment upgrades to handle pressure fluctuations safely
Improved monitoring and alarm systems to detect potential high-pressure situations
Validated procedures for responding to high-pressure events if they occur
A comprehensive risk assessment and mitigation plan related to pressure control in the manufacturing process
The Importance of Leak Prevention in Single-Use Systems
Single-use technologies have become increasingly prevalent in biopharmaceutical manufacturing due to their numerous advantages, including reduced risk of cross-contamination and increased flexibility. For all this to work, the integrity of these systems is paramount to ensure product quality and patient safety.
To address the challenges posed by leaks in single-use systems, manufacturers need to consider implementing a comprehensive control strategy. Here are some key approaches:
1. Integrity Testing
Implementing robust integrity testing protocols is crucial. Two non-destructive testing methods are particularly suitable for single-use systems:
Pressure-based tests: These tests can detect leaks by inflating components with air to a defined pressure. They can identify defects as small as 10 µm in flat bags and 100 µm in large-volume 3D systems.
Trace-gas-based tests: Typically using helium, these tests offer the highest level of sterility assurance and can detect even smaller defects.
2. Risk-Based Quality by Design (QbD) Approach
Single-use components and the manufacturing process must be established and maintained using a risk-based QbD approach that can help identify potential failure points and implement appropriate controls. This should include:
Comprehensive risk assessments
Validated procedures for responding to high-pressure events
Instead of using unapproved workarounds, companies need to develop and validate process modifications to prevent or mitigate high-pressure events. One thing to be extra cautious about is the worry of a temporary solution becoming a permanent one.
Conclusion
The Sanofi warning letter serves as a crucial reminder of the importance of maintaining the integrity of single-use systems in biopharmaceutical manufacturing. By implementing comprehensive control strategies, including robust integrity testing, risk-based approaches, and validated process modifications, manufacturers can significantly reduce the risk of leaks and ensure compliance with cGMP standards.
As the industry continues to embrace single-use technologies, it’s imperative that we remain vigilant in addressing these challenges to maintain product quality, patient safety, and regulatory compliance.
The recent FDA warning letter issued to Sanofi on January 15, 2025 highlights a critical issue that continues to plague pharmaceutical manufacturers – inadequate investigation of deviations. Specifically, the FDA cited Sanofi for “failure to thoroughly investigate any unexplained discrepancy or failure of a batch or any of its components to meet any of its specifications, whether or not the batch has already been distributed.”
This observation underscores the importance of robust deviation investigation and CAPA (Corrective and Preventive Action) systems.
The Importance of Thorough Investigations
Investigating deviations is not just a regulatory requirement – it’s a critical part of ensuring product quality and patient safety. The objective of an investigation is not merely to perform the investigation, but to improve the reliability of our manufacturing operations, the ultimate objective being increased quality and availability of those regulated healthcare products.
When companies fail to thoroughly investigate deviations, they miss opportunities to:
Consider both short-term corrections and long-term preventive measures
Assess potential risks of proposed CAPAs
Establish clear timelines and accountability for CAPA implementation
Conduct effectiveness checks to verify CAPA impact
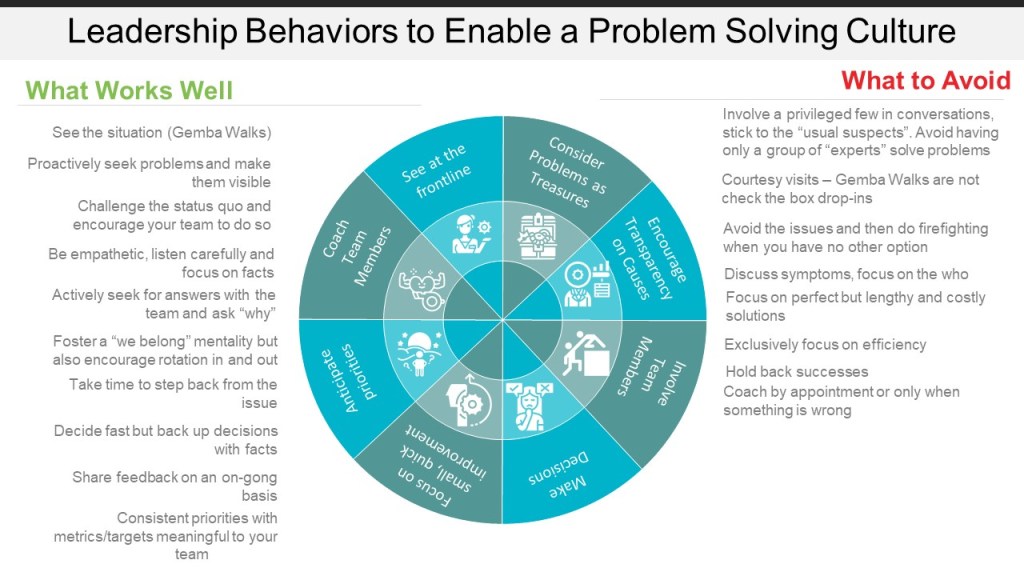
7. Foster a Culture of Quality
Management plays a critical role in creating an environment that supports thorough investigations.
Providing adequate time and resources for investigations
Encouraging open reporting of deviations without fear of blame
Recognizing and rewarding thorough investigation practices
Leading by example in prioritizing quality and patient safety
Common Pitfalls in Investigating Microbiological Contamination Events
When investigating microbiological contamination events there are often several pitfalls that can hinder the effectiveness of their investigations.
Inadequate Root Cause Analysis
One of the most significant pitfalls is failing to conduct a thorough root cause analysis. Investigators may be tempted to attribute contamination to superficial causes like “human error” without digging deeper into systemic issues. This shallow approach often leads to ineffective corrective actions that fail to prevent recurrence. Build in safeguards to avoid jumping to conclusion.
Overlooking Environmental Factors
Investigators sometimes neglect to consider the broader environmental context of contamination events. Factors such as air handling systems, water quality, and even compressed air can harbor contaminants. Failing to examine these potential sources may result in missed opportunities for identifying the true origin of contamination.
Insufficient Microbial Identification
Relying solely on phenotypic identification methods can lead to misidentification of contaminants. Phenotypic results can incorrectly point to laboratory contamination, while genotypic testing revealed a production-related issue. Using a combination of identification methods, including genotypic techniques, can provide more accurate and actionable results.
Premature Conclusion of Investigations
Pressure to close investigations quickly can lead to premature conclusions. This was evident in the Sanofi warning letter, where the FDA noted that investigations into critical deviations, including multiple microbiological contamination events, were inadequate. Rushing the process can result in overlooking important details and failing to implement effective corrective actions.
Failure to Consider Cross-Contamination
Investigators may not always consider the possibility of cross-contamination between products or areas within the facility. The presence of drug-resistant microbial contaminants, as observed in some studies, underscores the importance of examining potential routes of transmission and implementing strict hygiene procedures.
Inadequate Documentation
Poor documentation of investigation activities and rationale can undermine the credibility of findings and make it difficult to justify conclusions to regulators. The FDA’s warning letter to Sanofi highlighted this issue, noting that not all investigational activities were documented.
Neglecting Trending and Data Analysis
Failing to analyze contamination events in the context of historical data and trends can lead to missed patterns and recurring issues. Establishing and maintaining a comprehensive microflora database is essential for effective contamination control strategies and can provide valuable insights for investigations.
Insufficient Training of Investigators
Lack of properly trained and competent investigators can significantly impact the quality of contamination investigations. Ensuring that personnel have the necessary skills and knowledge to conduct thorough, science-based investigations is crucial for identifying true root causes and implementing effective corrective actions.
Conclusion
The Sanofi warning letter serves as a reminder of the critical importance of thorough deviation investigations in pharmaceutical manufacturing. By implementing robust investigation and CAPA processes, companies can not only avoid regulatory action but also drive continuous improvement in their operations. This requires ongoing commitment to developing investigator competencies, using structured methods, looking beyond superficial causes, and fostering a culture that values quality and learning from deviations.
As the industry continues to evolve, effective investigation practices will be essential for ensuring product quality, patient safety, and regulatory compliance. By viewing deviations not as failures but as opportunities for improvement, pharmaceutical manufacturers can build more resilient and reliable production systems.
The recent Sanofi Warning Letter certainly gets me thinking about the work of a consent decree and the scale and ‘stickiness‘ within an organization.
Scale of Remediation
In the Sanofi-Genzyme consent decree there were these concentric circles of required activities. At the center was the plant the issue was discovered, the Allston Landing Facility, which had the full brunt of remediation.
The next level out were the plants in Framingham and Northborough. They had remediation actions to be done, including reduced third party oversight for critical activities for a more limited time. The consent decree was on a much reduced scale at these sites.
The next level out was the former Genzyme sites beyond the Massachusetts core. They did alignment to the new standards created as part of the consent decree. Finally the rest of Sanofi, after Sanofi bought Genzyme, pretty much ignored it.
This balkanization meant that the culture across the organization never really changed. The cultural resistance of the site/silos fostered a culture of “us vs. them” mentality within the organization. Without a unified organizational culture, it is much harder to implement and maintain changes across the entire company.
The Slippery Slope: How Quality Improvements Can Erode Over Time
The erosion of quality culture at Sanofi demonstrated by this new Warning Letter isn’t unique to this case. Even when quality improvement initiatives are launched with great enthusiasm and initial success it is not uncommon for these hard-won gains to gradually erode over time, leaving organizations back where they started or even worse off. This phenomenon of “quality backsliding” can be frustrating and costly.
Why Quality Improvements Fade
There are several reasons why quality improvements may deteriorate over time:
Leadership Changes: When key champions of quality initiatives leave or change roles, their successors may not prioritize maintaining those improvements. New leaders often want to make their own mark, potentially abandoning or de-emphasizing existing quality programs.
Budget Cuts: In times of financial pressure, quality improvement efforts are often seen as “nice to have” rather than essential. Resources dedicated to sustaining improvements may be reallocated, leading to a gradual decline in performance.
Complacency: Initial success can breed complacency. Once targets are met, there may be less motivation to continue pushing for further improvements or even maintaining current standards.
Loss of Focus: As new priorities emerge, attention and resources can shift away from quality initiatives. Without ongoing commitment, processes can slowly revert to old, less effective ways of working.
Lack of Standardization: If improvements aren’t fully standardized and integrated into daily operations, they remain dependent on individual efforts rather than becoming part of the organizational culture.
I think we will be evaluating the Sanofi Warning Letter of January 15th, 2025 for a while. Received at the Framingham manufacturing site, this Warning Letter will fuel case studies about the pendulum of compliance and how it can swing perhaps a bit too erratically.
This site is the sister site to the former Genzyme site (last time I checked owned by Resilience and mothballed) in Allston, MA.
The Genzyme consent decree was a significant regulatory action taken by the U.S. Food and Drug Administration (FDA) in response to ongoing manufacturing quality issues at Genzyme’s Allston Landing facility in Massachusetts. Here’s a chronological overview of the key events:
In October 2008, an FDA inspection of the Allston plant led to the issuance of an FDA Form 483, highlighting various deficiencies. In February 2009, the FDA issued a Warning Letter to Genzyme, detailing issues with microbiological contamination control procedures and bioburden monitoring. Then in June 2009, Genzyme detected a virus in one of its bioreactors, leading to a six-week production interruption and subsequent drug shortages. A follow-up FDA inspection in November 2009 revealed ongoing significant problems, resulting in a 49-item Form 483.
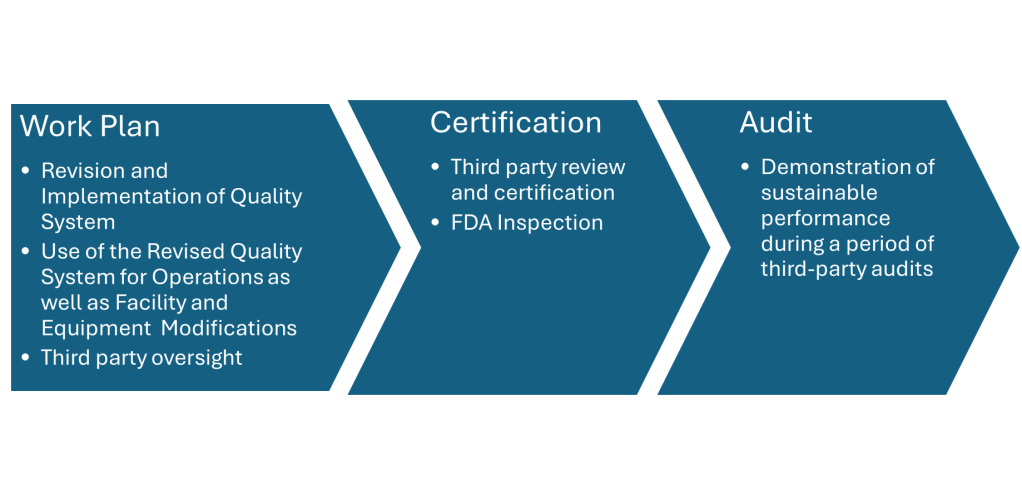
On May 24, 2010, Genzyme signed a consent decree with the FDA. The consent decree required Genzyme to adhere to a strict timetable to bring the Allston plant into compliance with FDA regulations.
The next few years a comprehensive remediation plan was implemented with ongoing oversight from a third-party consultant. The company then went through a certification process, FDA inspection, and surveillance by a third party for another five years before being able to request an end to the consent decree.
I believe when Sanofi sold the Allston site to Resilience (Sanofi bought Genzyme in 2011), the consent decree had pretty much finished that surveillance period, but I can find no evidence of the company petitioning the court to lift the consent decree. So I have no idea what that means.
The Framingham sites (which this Warning Letter is addressed to) here under the consent decree but to a lesser amount of oversight. So to see this new Warning Letter, for the new construction done in the mid 2010s is pretty sad for me.
There’s a lot to unpack here that is relevant to SUS biologics manufacturing facilities, but that will be a future post. I need to go get a drink.
Occasionally the FDA writes a Warning Letter that is a succinct lesson in what is important about a quality system. In this Warning Letter they masterfully describe what a CAPA Program is. As this one further describes how to deal with an inadequate cleaning program it is near and dear to my heart.