In a regulated industry, such as pharmaceuticals or medical devices, knowing your changes impact your regulatory partners is a critical aspect of change management. For example, the MHRA in their yearly summarizations of GMP inspection deficiencies consistently cites failure to perform adequate review of need of regulatory notification (for example, see 2016 trends). And to be frank, we in the industry are often looking for more guidance, which drives responses like ICHQ12 and the FDA’s March 2018 draft guidance CMC Changes to an Approved Application: Certain Biological Products and all the other similar guidances out there.
These all follow a similar risk-based approach, and this approach should be built into your change management system (and applicable change control process).
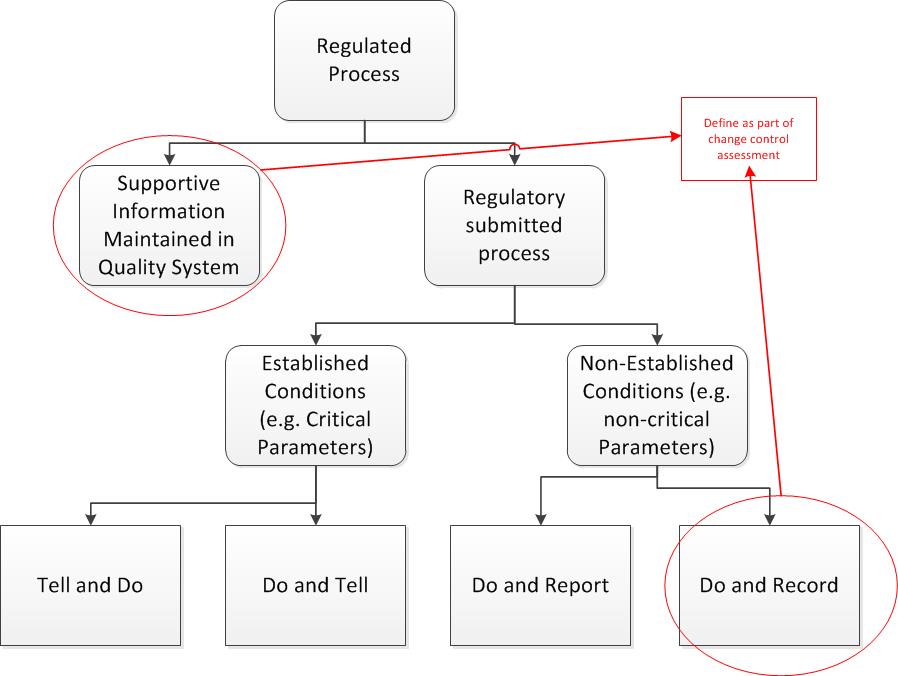
The major difference between Supportive Information and Do-and-Record is usually what goes in your product quality report (APR/PQR). Fro example, I often see qualification of facility fit into the Do-and-Record area. These changes may not be fillable, but you certainly want to review and account for.
Many companies manage this through their regulatory affairs organization, but that can be time consuming. It is better to take the time to identify the supportive and do-and-record categories out front, thus removing the need for an extra assessment. The PQR review process is a great tool for ensuring consistent execution.
This risk based approach should look at the dossiers, taking into account any special market considerations, as well as current best practices in the regulations. For those companies lucky enough to be more towards the QbD model, established conditions will greatly help here.
Then build a matrix to help guide your changes. An example could include items like these:
| Facility, Equipment, Manufacturing Systems, Utilities & Automation | Equipment/instrument maintenance |
| Decommissioning of equipment not classified as critical equipment | |
| Computer programming that affects non-production equipment | |
| Alarms (i.e., notification system for out of tolerances) | |
| Cleaning and Sanitization of Manufacturing facilities and non-product Contact equipment | |
| Upgrade of Application Software or operating system | |
| Alarm setpoint changes | |
| Creating user groups and modifying user group privileges | |
| Tuning parameter, adjustment to the gain, reset and rate of a PID controller | |
| Manufacturing Processes | In-process labeling |
| Changes to Process Control and Operating Parameters (tightening/shifting) within current non-established conditions | |
| Change in equipment sterilization times | |
| The addition of in-process or final product samples | |
| Changes to sample volume for in-process or finished product samples | |
| Addition of new ancillary equipment (e.g. no product contact, does not control process steps) to the process |
You can then further delineate between Supportive Information and Do-and-Record on a few other criteria, such as qualification/validation impact.
Like many areas of good system management, this is an area where a forethought and design can reap dividends in making your changes more nimble while preventing a compliance mishap. Tapping into the PQR makes all this part of your knowledge management system, and allows you to grow as your needs grow. This is definitely not a once-and-done process.

9 thoughts on “Regulatory Impact of Changes”