There has been increasing evidence in recent years that research in life sciences is lacking in reproducibility and data quality. This raises the need for effective systems to improve data integrity in the evolving non-GxP research environment. Reproducibility is a defining principle of scientific research, and broadly refers to the ability of researchers, other than the original researchers, to achieve the same findings using the same data and analysis data reproducibility is key to the reinforcement and credibility of scientific evidence. All results should be replicable by different investigators in varied geographical settings, using independent data, instruments, and analytical methods.
Some examples:
In 2022 there were 11 Federal Register notices with ORI findings of research misconduct that involved Public Health Service support or funding. These cases included falsified data submitted in National Institutes of Health grant applications and PHS-supported publications. These cases resulted debarment periods of up to four years and supervision periods of up to 12 years.
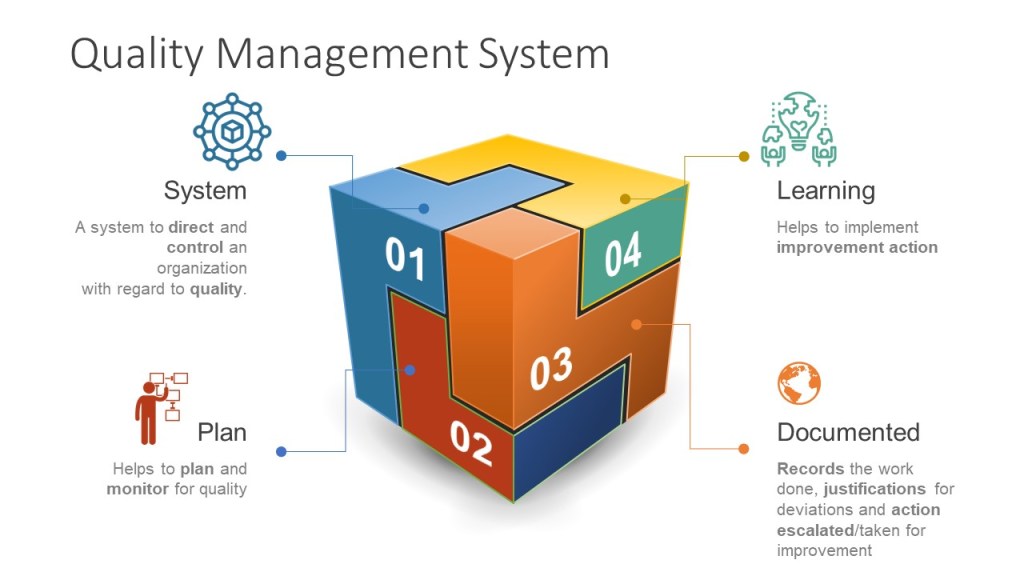
Without a doubt it is critical to build a quality culturewithin our research organizations. Through educating our scientific staff we can continue to innovate and discover new pathways, new drugs and new treatments. Efficient processes enhance research effectiveness and lead to scientific discoveries. Data integrity supports good science, drug safety, products and treatment development for patients and customers. While this looks similar in research as in later phases there are 4 primary pillars:
Train researchers on basic documentation processes and good scientific practices to ensure data integrity and quality. Targeted training should be added on new guidelines, processes and regulations applied to their specific activities.
Bespalov, A., Bernard, R., Gilis, A., Gerlach, B., Guillén, J., Castagné, V., Lefevre, I. A., Ducrey, F., Monk, L., Bongiovanni, S., Altevogt, B., Arroyo-Araujo, M., Bikovski, L., Bruin, N. de, Castaños-Vélez, E., Dityatev, A., Emmerich, C. H., Fares, R., Ferland-Beckham, C., … Steckler, T. (2021, May 24). Introduction to the EQIPD Quality System. eLife. https://elifesciences.org/articles/63294
There is no term more misused and misunderstood than “Phase Appropriate.” It is one of those terms that just about everyone involved in FDA-regulated industries has an opinion on and one where we all get tripped up.
What do we mean by phase?
Drug development can be divided into discovery, preclinical studies, clinical development, and market approval.
Each one of these phases is further broken down.
It is also important to remember that certain activities may start in earlier phases. For example, for manufacturing, tech transfer, and commercial manufacturing can start in Phase 3 (and more and more these days even 2!).
An investigational drug for use in a phase 1 study, as described in § 312.21(a) of this chapter, is subject to the statutory requirements set forth in 21 U.S.C. 351(a)(2)(B). The production of such drug is exempt from compliance with the regulations in part 211 of this chapter. However, this exemption does not apply to an investigational drug for use in a phase 1 study once the investigational drug has been made available for use by or for the sponsor in a phase 2 or phase 3 study, as described in § 312.21(b) and (c) of this chapter, or the drug has been lawfully marketed. If the investigational drug has been made available in a phase 2 or phase 3 study or the drug has been lawfully marketed, the drug for use in the phase 1 study must comply with part 211.
Guideline on the responsibilities of the sponsor with regard to handling and shipping of investigational medicinal products for human use in accordance with Good Clinical Practice and Good Manufacturing Practice
Processes characterized and Production and Process Controls (PPC) identified
Analytical methods are qualified
Materials acceptance criteria
Critical vendors qualified
Phase 3:
Processes validated with Production and Process Controls (PPC) identified and controlled
Validation of analytical methods
Materials have been fully qualified and tested upon receipt as appropriate
What About the Quality System?
ICH Q10 clearly spells out the PQS requirements, breaking down into stages of Pharmaceutical Development (usually Phase 1 and earlier), Technology Transfer (usually phase 2), Commercial Manufacturing (which may start before approval) and Product Discontinuation. Q10 then lays out the expectations by these stages for the four key elements of:
Process performance and product quality monitoring system
Corrective action and preventive action (CAPA) system
Change management system
Management review of process performance and product quality.
Pharmaceutical Development
Technology Transfer
Commercial Manufacturing
Product Discontinuation
Process Performance and Product Quality
Process and product knowledge generated and process and product monitoring conducted throughout development can be used to establish a control strategy for manufacturing.
Monitoring during scale-up activities can provide a preliminary indication of process performance and the successful integration into manufacturing. Knowledge obtained during transfer and scale up activities can be useful in further developing the control strategy.
A well-defined system for process performance and product quality monitoring should be applied to assure performance within a state of control and to identify improvement areas.
Once manufacturing ceases, monitoring such as stability testing should continue to completion of the studies. Appropriate action on marketed product should continue to be executed according to regional regulations.
Corrective Action and Preventive Action
Product or process variability is explored. CAPA methodology is useful where corrective actions and preventive actions are incorporated into the iterative design and development process.
CAPA can be used as an effective system for feedback, feedforward and continual improvement.
CAPA should be used and the effectiveness of the actions should be evaluated.
CAPA should continue after the product is discontinued. The impact on product remaining on the market should be considered as well as other products which might be impacted.
Change Management
Change is an inherent part of the development process and should be documented; the formality of the change management process should be consistent with the stage of pharmaceutical development.
The change management system should provide management and documentation of adjustments made to the process during technology transfer activities.
A formal change management system should be in place for commercial manufacturing. Oversight by the quality unit should provide assurance of appropriate science and risk based assessments.
Any changes after product discontinuation should go through an appropriate change management system.
Management Review of Process Performance and Product Quality
Aspects of management review can be performed to ensure adequacy of the product and process design.
Aspects of management review should be performed to ensure the developed product and process can be manufactured at commercial scale.
Management review should be a structured system, as described above, and should support continual improvement.
Management review should include such items as product stability and product quality complaints.
ICH Stage appropriate quality system elements
Together with ICH Q9, this sets forth a framework of building knowledge and risk management into all aspects of the system together with a robust issue management mindset. There are really three things driving this.
Consistency in execution
Document decision making
Follow through
Some aspects remain pretty steady in all phases/stages, while others will grow as the organization develops.
The Difference Between Maturity and Phase Appropriate
People confuse phase appropriate with maturity all the time. Phase appropriate means doing the right activities in the right order. Maturity means the how is the most effective possible.
Quality Management Maturity (QMM) is the state attained when drug manufacturers have consistent, reliable, and robust business processes to achieve quality objectives and promote continual improvement. This is both composed of phase independent and phase dependent aspects.
Remember, a Quality Culture is the foundation that makes the rest of this happen.
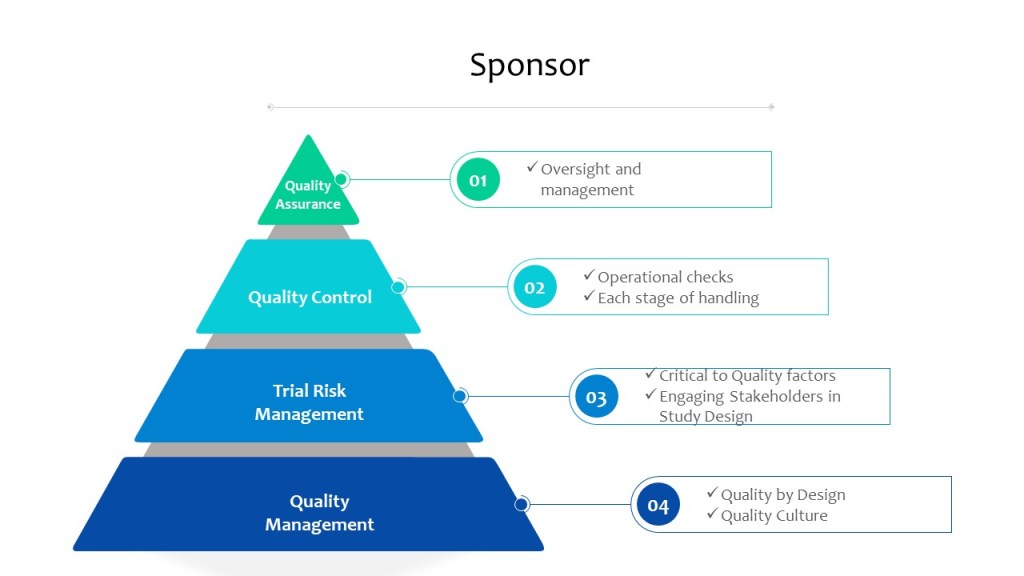
With ICH E6(r3) in draft, I think it is important to look at the current state of risk management expectations in a clinical study.
Risk management is an essential part of any clinical study, and is a critical component of the ICH E6 and E8 guidelines for Good Clinical Practice (GCP). These guidelines provide a framework for ensuring the safety and well-being of study participants, as well as the integrity and reliability of the study data. By following the principles outlined in these guidelines, researchers can help to ensure that their study results are reliable and can be used to inform clinical practice.
Through risk management we ensure the four main goals of the GCPs are obtained.
The ICH E6 guideline provides recommendations for the conduct of clinical trials, emphasizing the importance of risk management, specifying that a risk management plan should be developed and implemented for each study. The guideline also provides recommendations for the content of the risk management plan, including the identification of potential risks, the assessment of their likelihood and potential impact, and the development of strategies for managing or mitigating those risks.
Risk management is a key enabler and result of the quality management system.
The ICH E8 guideline, which focuses on the conduct of clinical trials also emphasizes the importance of risk management. It specifies that the risk management plan should include a comprehensive evaluation of the risks associated with the study interventions, as well as a plan for managing or mitigating those risks. The guideline also recommends that the risk management plan be regularly reviewed and updated as needed, to ensure that it continues to effectively address the risks facing the study.
When planning a clinical study, sponsors must carefully consider the potential risks involved and take steps to minimize them. Sources of the risk assessment include performing a thorough literature review to identify any known risks associated with the study interventions, as well as conducting pre-study assessments to identify potential risks specific to the study population. E8 also state sthe importance of a wide variety of stakeholders, including the patient population.
Once the study is underway, it’s important to closely monitor for potential risks and have a plan in place for managing them.
In addition to protecting the safety of study participants, effective risk management is also essential for maintaining the integrity of the data being collected. Risks to the study data might include things like errors in data entry or missing data, which can compromise the validity of the study results. To address these risks, sponsors must have robust quality control measures in place, such as regular data audits and checks for missing or inconsistent data.
Overall, the role of risk management in a clinical study is to ensure the safety and well-being of study participants, while also protecting the integrity of the data being collected. By carefully considering and managing potential risks, researchers can help to ensure that their study results are reliable and can be used to inform clinical practice.
Risk Based Monitoring
Risk-based monitoring is a approach to monitoring the quality of a clinical study that focuses on identifying and addressing potential risks to the study. This approach involves regularly assessing the risks associated with a study and implementing strategies to manage or mitigate those risks.
In a risk-based monitoring approach, the study team typically uses a risk register to identify and assess potential risks to the study, such as the potential for errors in data collection or analysis, or the potential for adverse events in study participants. The team then develops a plan for addressing these risks, which might involve implementing additional quality control measures or training for study staff.
During the study, the team regularly monitors for potential risks and takes action to address them as needed. This might involve conducting regular audits or reviews of the study data to identify potential errors, or monitoring the health and well-being of study participants to identify and address any adverse events.
Overall, the goal of risk-based monitoring is to ensure the quality and integrity of a clinical study by proactively identifying and addressing potential risks. By using a risk-based approach, the study team can help to ensure that the study results are reliable and can be used to inform clinical practice.
Risk Register
A risk register is a document that is used to identify, assess, and track potential risks in a clinical study. It typically includes a list of identified risks, along with information about their likelihood and potential impact, as well as the actions that are being taken to manage or mitigate the risks.
In a clinical study, a risk register might include risks such as the potential for errors in data collection or analysis, the potential for adverse events in study participants, or the potential for the study to be impacted by external factors, such as changes in regulatory requirements.
The purpose of a risk register in a clinical study is to help the study team identify and prioritize potential risks, and to develop strategies for addressing them. By having a clear and comprehensive overview of the risks that a study is facing, the team can take proactive steps to manage or mitigate those risks, and can monitor their progress over time.
Overall, a risk register is an essential tool for managing risks in a clinical study. By providing a clear and comprehensive overview of potential risks, it helps the study team identify and address risks in a proactive and effective way.
Identifying potential risks: The first step in implementing a clinical risk management program is to identify potential risks to the study, such as the potential for errors in data collection or analysis, or the potential for adverse events in study participants. This might involve reviewing the study protocol and data collection tools, consulting with the study team and other stakeholders, and conducting a thorough assessment of the study environment.
Assessing risks: Once potential risks have been identified, the next step is to assess their likelihood and potential impact. This will help to prioritize the risks and determine the appropriate level of response. For example, a risk with a high likelihood and a high potential impact might require more immediate action, while a risk with a low likelihood and a low potential impact might not require as much attention.
Developing strategies for managing risks: Based on the assessment of risks, the next step is to develop strategies for managing or mitigating those risks. This might involve implementing additional quality control measures, providing training to study staff, or conducting regular audits or reviews of the study data. The goal is to develop a comprehensive and effective plan for addressing the identified risks.
Monitoring for potential risks: Once the risk management plan is in place, it’s important to regularly monitor for potential risks and take action to address them as needed. This might involve conducting regular audits or reviews of the study data, or monitoring the health and well-being of study participants. By proactively monitoring for potential risks, the study team can help to ensure the safety and well-being of study participants, as well as the integrity and reliability of the study data.
Follow-up and corrective action: If potential risks are identified during the study, it’s important to take prompt action to address them. This might involve implementing corrective action plans, such as retraining study staff or revising the study protocol. It’s also important to track the progress of these plans and ensure that they are effective in addressing the identified risks. By taking timely and effective action to address potential risks, the study team can help to ensure the safety and well-being of study participants, as well as the integrity and reliability of the study data.
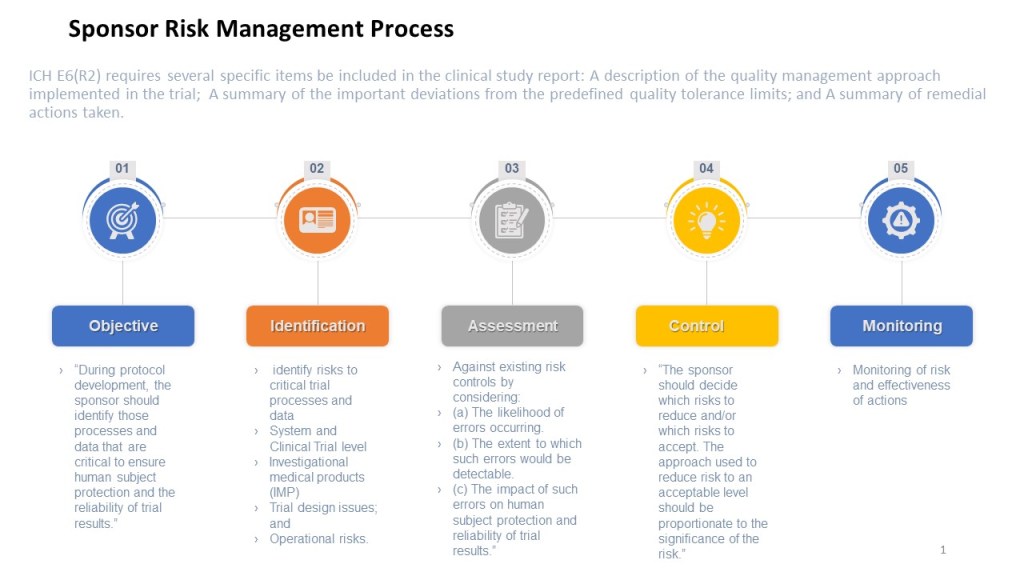
Risk Management in the Clinical Study Process
To summarize, each clinical study should:
Identify Risks
Before the study begins, the sponsor should perform a thorough review of the study protocol, data collection tools, and other study-related documents to identify potential risks to the study.
The cross-functional study team, CROs and other relevant stakeholders, such as the sponsor and regulatory authorities, to identify additional potential risks.
All identified risks should be documented in the study’s risk register.
2. Assess Risks
For each identified risk, assess its likelihood and potential impact on the study.
The risks should be prioritized based on their likelihood and potential impact, with a focus on the highest-priority risks.
3. Manage Risks
For each identified risk, the sponsor should develop a plan for managing or mitigating the risk. This plan should be documented in the study’s risk register.
The plan for managing or mitigating each risk should include specific actions to be taken, as well as the individuals or groups responsible for implementing those actions.
4. Monitor Risks
Regularly monitor key risk indicators and the study for success of the study risk plan and to identify new potential risks and take action to address them as needed. This might involve conducting regular audits or reviews of the study data, or monitoring the health and well-being of study participants.
Any significant risks that arise during the study should be reported to the sponsor and relevant regulatory authorities.
Automation has been a truism throughout my career. Organizations that leveraged that automation to create value were superior to the ones which used that automation as an excuse to cut jobs.
As we move oh so quickly to dealing with the impact of hyper-automation on our organizations it is important to have a vision and a strategy. Apply quality principles, and remember to drive out fear through the strategic execution.
The word quality is a loaded word in organizations, and I’m sure most of my readers have been in a least one major discussion that has felt like an Abbott and Costello routine.
The difference between “quality” and the “Quality department” is that “quality” refers to the overall level of excellence or excellence in a product, service, or process, while the “Quality department” is part of an organization that is responsible for ensuring that the organization’s products, services, or processes meet certain quality standards.
In other words, “quality” is a general concept that refers to the level of excellence or excellence in something, while the “Quality department” is a specific part of an organization that is responsible for managing and improving the quality of that organization’s products, services, or processes.
The Quality department typically plays a key role in ensuring that an organization’s products, services, or processes meet the required quality standards. This might involve activities such as conducting quality assurance audits, implementing quality control measures, or providing training and support to help employees understand and comply with quality standards.
Overall, the difference between “quality” and the “Quality department” is that “quality” is a broad concept that refers to the overall level of excellence or excellence in something, while the “Quality department” is the specific part of an organization that is responsible for managing and improving the quality of that organization’s products, services, or processes.
In FDA-regulated industries, this continues to be a stressful point. We have some regulations that specifically call out the Quality Unit or Quality Control (a different point of fun), while others provide quality expectations that may or may not be the responsibility of the Quality Unit, depending on the way your organization is built.
Add to this that quality is a culturally sensitive term. It gets to the heart of what people consider integral to themselves. That they have quality in their work. And there can be gaps between people’s perceptions and the reality of the organization. The whole concept of what quality is in an organization gets to three central aspects:
Role Conception: what people think their jobs are and how they have been trained to perform them
Role Expectation: what others in the organization think another person’s job is and how it should be carried out
Role Behavior: what people actually do in carrying out their job
So we have quality as a set of habits and practices and Quality as a concept of a role within an organization. And the boundaries between the two can be contentious. Add in the quality control layer (and how quality control does not require a department called quality control) and we can have a whole fun set of arguments.
This post was brought to you by me being in a meeting where someone referenced a version of the golden triangle and I instantly wondered what work someone else was trying to foist off onto me.