Guidances are an interesting part of our job. As a best practice, they show one way to get to the desired end result, but there can be other ways but the presence of guidance can obscure those possibilities. Often times if an agency goes to the level of detail to show you what good looks like you’d be foolish not to try to meet them there. Other times guidance can be a real head scratcher.
Good article, and of interest to the non-lawyers like myself who have to live within the boundaries.
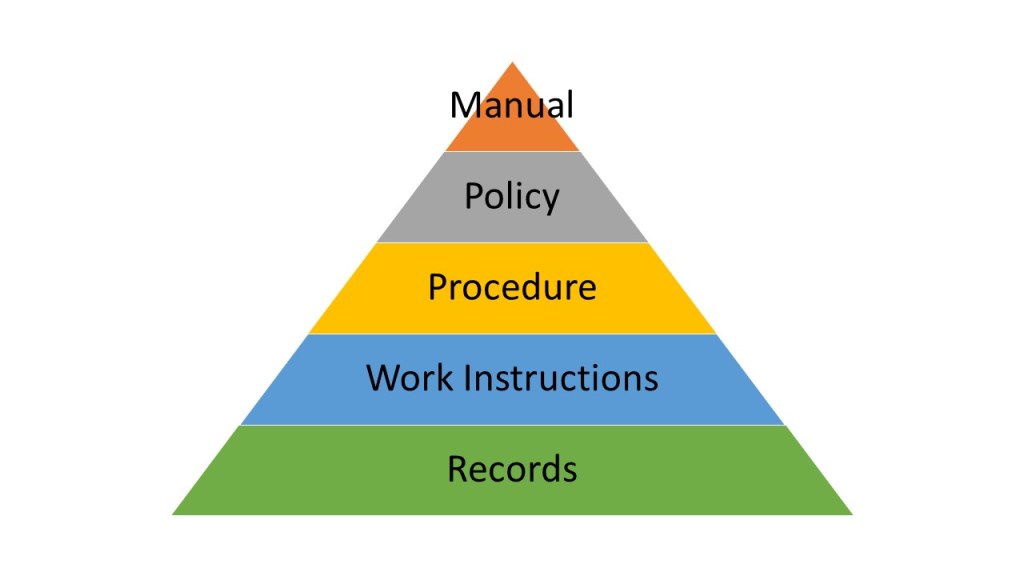
A fairly traditional document hierarchy, in line with ISO 9001 and other standards looks like this:
Document hierarchy
This process tends to support best an approach where there is a policy that states requirements to do X, a procedure that gives the who, what, when of X, and work instructions that provide the how of X, which results in a lot of records providing X was done.
But life is complicated, and there are sets of activities that combine the Xs in a wide variety, and in complicated environments there may be multiple ways to bundle the Xs.
This is why I add a layer between policy and procedure, called the program, which is a mapping requirement that shows the various ways to interpret the requirements to specific needs.
Document hierarchy with Programs
The program document level shouldn’t be a stranger to those in the GMP world, ICH Q11 control strategy and the Annex 1 contamination control strategy are two good examples. What this document does is tie together processes and demonstrates the design that went into it.
The beauty of this document is that it helps translate down from the requirements (internal and external) to the process and procedures (including technology), how they interact, and how they are supported by technical assessments, risk management, and other control activities. Think of it as the design document and the connective tissue.
There has been a lot of press lately for the Abbott Nutrition recall of infant formula. Fundamentally this is a colossal failure of our regulatory program, another failure in a long string of failures, and confirmation that the time is now for radical changes in the agency.
The optimist in me hopes that this calamity will drive needed change, as has been the unfortunate history of regulatory change in this country. I’m just not sure I hold enough confidence in Congress to get the job done.
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Chapter 3: Premises and Equipment, (2014)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Chapter 5: Production, (2014)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Part II: Basic Requirements for Active Substances used as Starting Materials, (2014)
European Union, Guidelines of 19 March 2015 on the formalized risk assessment for ascertaining the appropriate good manufacturing practice for excipients of medicinal products for human use, Official Journal of the European Union, (2015/C 95/02), (2015)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Annex 2: Manufacture of Biological active substances and Medicinal Products for Human Use, (2018)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Annex 3 Manufacture of Radiopharmaceuticals, (2008)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Annex 14 Manufacture of Medicinal Products Derived from Human Blood or Plasma, (2011)
European Commission, EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products, (2017)
European Union, Guidelines of 5 November 2013 on Good Distribution Practice of medicinal products for human use, Official Journal of the European Union, (2013/C 343/01), (2013),
European Union, Guidelines of 19 March 2015 on principles of Good Distribution Practice of active substances for medicinal products for human use, Official Journal of the European Union, (2015/C 95/01), (2015)
EMA Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities (20 November 2014)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, subpart C = Building and Facilities, sec. 211.42 Design and construction features (b), (c)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart F – Production and Process Controls, sec. 211.113 Control of microbial contamination (a), (b)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart B – Organization and Personnel, sec.211.28 Personnel responsibilities (a)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart E – Control ofComponents and Drug Product Containers and Closures, sec. 211.80 General requirements. (b)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart E – Control of Components and Drug Product Containers and Closures, sec. 211.84 Testing and approval or rejection of components, drug product containers, and closures (d)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart D – Equipment, sec.211.67 Equipment cleaning and maintenance (a)
U.S. Food & Drug Administration, Code of Federal Regulation Title 21, part 211 current good manufacturing practice for finished pharmaceuticals, Subpart C – Buildings and Facilities, sec. 211.56 Sanitation (c)
U.S. Food & Drug Administration, Guidance for Industry Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice, (2004)
U.S. Food & Drug Administration, Guidance for Industry – Good Manufacturing Practice Considerations for Responding to COVID-19 Infection in Employees in Drug and Biological Products Manufacturing, (2020)
U.S. Food & Drug Administration, Guidance for Industry – Guidance for Industry Non-Penicillin Beta-Lactam Drugs: A CGMP Framework for Preventing Cross Contamination, (2013)
U.S. Food & Drug Administrationn, Guidance for Industry Current Good Manufacturing Practice—Guidance for Human Drug Compounding Outsourcing Facilities Under Section 503B of the FD&C Act, Draft Guidance. https://www.fda.gov/media/88905/download (accessed Mar 6, 2022)
Pharmaceutical Inspection Co-operation Scheme gmp guide, 2nd targeted consultation document on revision of annex 1
Pharmaceutical Inspection Co-operation Schemepharmaceutical inspection co-operation scheme gmp guide, ps inf 25 2019 (rev. 1) draft, manufacture of advanced therapy medicinal products for human use
Pharmaceutical Inspection Co-operation Scheme gmp guide, ps inf 26 2019 (rev. 1) draft, manufacture of biological medicinal substances and products for human use
Pharmaceutical Inspection Co-operation Scheme gmp guide, pe 009-15 (part i), guide to good manufacturing practice for medicinal products part i
Pharmaceutical Inspection Co-operation Scheme gmp guide, pe 009-15 (part ii), guide to good manufacturing practice for medicinal products part ii
Pharmaceutical Inspection Co-operation Scheme gmp guide, pe 009-15 (annexes), guide to good manufacturing practice for medicinal products annexes
World Health Organisation, good manufacturing practices for pharmaceutical products: main principles, annex 2, who technical report series 986, 2014,
World Health Organisation, who good manufacturing practices for active pharmaceutical ingredients (bulk drug substances), annex 2, who technical report series 957, 2010
World Health Organisation, points to consider for manufacturers and inspectors: environmental aspects of manufacturing for the prevention of antimicrobial resistance annex 6, who technical report series 1025, 2020
World Health Organisation, WHO good manufacturing practices for sterile pharmaceutical products, annex 6, who technical report series 961, 2011
World Health Organisation, WHO good manufacturing practices for biological products, annex 3, who technical report series 996, 2016
World Health Organisation, WHO good manufacturing practices for the manufacture of investigational pharmaceutical products for clinical trials in humans, annex 7, who technical report series 863, 1996
World Health Organisation, WHO good manufacturing practices for radiopharmaceutical products annex 2, who technical report series 1025, 2020
World Health Organisation, WHO GMP for Pharmaceutical Products containing Hazardous Substances, TRS 957, Annex-3 (2010)
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human use, Quality Risk Management, Q8 (R2), Pharmaceutical Development, August 2009. https://database.ich.org/sites/default/files/Q8%28R2%29%20Guideline.pdf (Accessed Mar 06, 2022)
Quality as a profession is often put into the position of being the cop or gatekeeper. There are a set of regulations and standards that must be met, and it can be easy, especially early in one’s career and without proper mentoring, to start to see absolutes.
Compromise is not a weakness in a quality professional, it is a strength.
There are times when, instead of ramping up your argment fill fore to make a case, it is better to step back and think about where you can comprise and still convince the organization to implement most, if not all, of your ideas.
This is where the change accelerators come in. Articulate the vision, and then utilize compromise the build and evolve the guiding coalition and turn that into an army of the willing.
Pilot programs, soft launches, workshops. These tools will help you find your allies and facilitate a solution.
Part of comprise is knowing what you can and will settle for. These questions can help:
What is the first thing I am willing to cede? It may be the timeline or a small adoption of your solution, such as a pilot project.
What is my backup plan? If the stakeholders don’t adopt my plan but offer a counterproposal, what am I willing to accept and jump on board with?
What is fueling the stakeholders’ reluctance? Ask questions, engage in “yes…but…and” practice.
Can I rework my argument? Is there an opportunity to come back with a revised pitch? Can you simplify or emphasize specific parts of your argument? Can you break it down into smaller parts – such as building blocks – first gaining support for the concept, ten gaining support for the first step to test its success, and then building support for the next step or phase?
Compromise is negotiation, and it requires all your emotional intelligence skills – patience, active listening, respect for the stakeholders’ position.
Have a vision, a plan, can really help. You will never get to 100% of meeting a requirement but being able to articulate what great looks like and then showing a plan that builds at a good clip, that allows compromise, will allow you to make continued progress and adjust as you go. Your systems will be stronger as a result.