Predicts compliance with FDA 21 CFR 211.100 (process control)
FDA 21 CFR 211, ICH Q10, ICH Q9
Lagging
Average Time to Close Change Requests
Validates efficiency of change implementation (EudraLex Annex 15)
EU GMP Annex 15
KRI
Leading
Unresolved CAPAs Linked to Change Requests
Identifies systemic risks before deviations occur (FDA Warning Letters)
21 CFR 211.22, ICH Q7
Lagging
Repeat Deviations Post-Change
Reflects failure to address root causes (FDA 483 Observations)
21 CFR 211.192
KBI
Leading
Cross-Functional Review Participation Rate
Encourages proactive collaboration in change evaluation
ICH Q10 Section 3.2.3
Lagging
Reduction in Documentation Errors Post-Training
Validates effectiveness of staff competency programs
EU 1252/2014 Article 14
Key Performance Indicators (KPIs)
Leading KPI:
Change Requests with Completed Risk Assessments: Measures proactive compliance with FDA requirements for risk-based change evaluation. A rate <90% triggers quality reviews.
Lagging KPI:
Time to Close Changes: Benchmarks against EMA’s 30-day resolution expectation for critical changes. Prolonged closure (>45 days) indicates process bottlenecks.
Key Risk Indicators (KRIs)
Leading KRI:
Unresolved CAPAs: Predicts validation gaps; >5 open CAPAs per change violates FDA’s “state of control” mandate.
Lagging KRI:
Repeat Deviations: >3 repeat deviations quarterly triggers mandatory revalidation per FDA 21 CFR 211.180.
Documentation Errors: Post-training error reduction <30% prompts requalification under EU GMP Chapter 4.
Implementation Guidance
Align with Regulatory Thresholds: Set leading KPI targets using FDA’s 2025 draft guidance: ≥95% risk assessment completion for high-impact changes.
Automate Tracking: Integrate metrics with eQMS software to monitor CAPA aging (leading KRI) and deviation trends (lagging KRI) in real time.
Link to Training: Tie lagging KBIs to annual GMP refresher courses, as required by EU 1252/2014 Article 14.
Why It Matters: Leading metrics enable proactive mitigation of change-related risks (e.g., unresolved CAPAs predicting audit failures), while lagging metrics validate adherence to FDA’s lifecycle approach for process validation. Balancing both ensures compliance with 21 CFR 211’s “state of control” mandate while fostering continuous improvement.
One of the many fascinating items in the recent Warning Letter to Sanofi is the FDA’s direction to provide a plan to perform “timely technological upgrades to the equipment/facility infrastructure.” This point drives home the point that staying current with technological advancements is crucial for maintaining compliance, improving efficiency, and ensuring product quality. Yet, I think it is fair to say we rarely see it this bluntly put as a requirement.
One of the many reasons this Warning Letter stands out is that this is (as far as I can tell) the same facility that won the ISPE’s Facility of the Year award in 2020. This means it is still a pretty new facility, and since it is one of the templates that many single-use biotech manufacturing facilities are based on, we had best pay attention. If a failure to maintain a state-of-the-art facility can contribute to this sort of Warning Letter, then many companies had best be paying close attention. There is a lot to unpack and learn here.
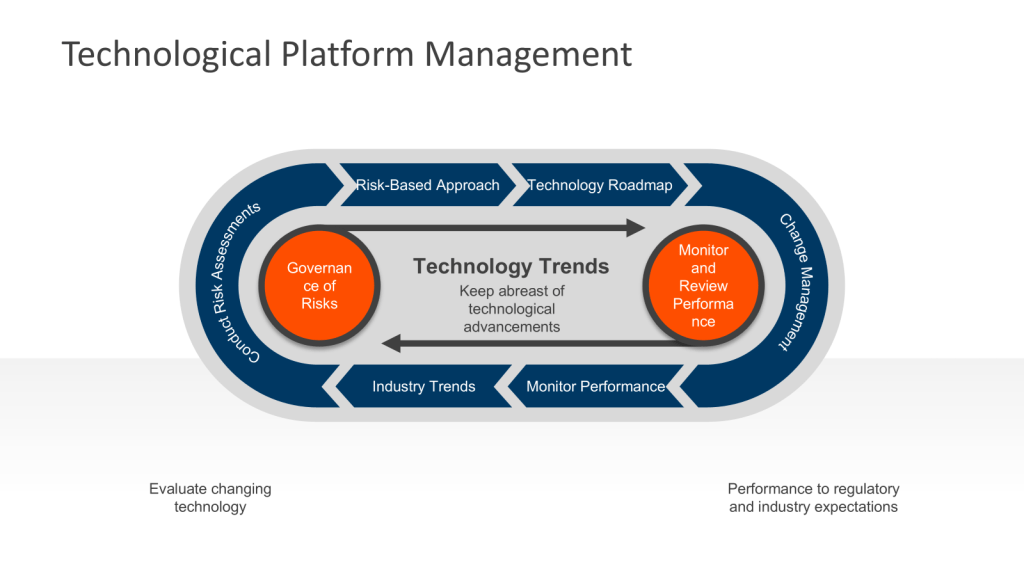
Establishing an Ongoing Technology Platform Process
To meet regulatory requirements and industry standards, facilities should implement a systematic approach to technological upgrades.
1. Conduct Regular Assessments
At least annually, perform comprehensive evaluations of your facility’s equipment, systems, and processes. This assessment should include:
Prioritize upgrades based on their potential impact on product quality, patient safety, and regulatory compliance. Utilize living risk assessments to get a sense of where issues are developing. These should be the evolution of the risk management that built the facility.
4. Create a Technology Roadmap
Develop a long-term plan for implementing upgrades, considering:
Budget constraints and return on investment
Regulatory timelines for submissions and approvals
Production schedules and potential downtime
Integration with existing systems and processes
5. Implement Change Management Procedures
Ensure there is a robust change management process in place to ensure that upgrades are implemented safely and effectively. This should include:
Detailed documentation of proposed changes
Impact assessments on product quality and regulatory compliance
6. Appropriate Verification – Commissioning, Qualification and Validation
Conduct thorough verification activities to demonstrate that the upgraded equipment or systems meet predetermined specifications and regulatory requirements.
7. Monitor and Review Performance
Continuously monitor the performance of upgraded systems and equipment to ensure they meet expectations and comply with cGMP requirements. Conduct periodic reviews to identify any necessary adjustments or further improvements. This is all part of Stage 3 of the FDA’s process validation model focusing on ongoing assurance that the process remains in a state of control during routine commercial manufacture. This stage is designed to:
Anticipate and prevent issues before they occur
Detect unplanned deviations from the process
Identify and correct problems
Leveraging Advanced Technologies
To stay ahead of regulatory expectations and industry trends, consider incorporating advanced technologies into your upgrade plans:
Single-Use Systems (SUS): Implement disposable components to reduce cleaning and validation requirements while improving flexibility.
Modern Microbial Methods (MMM): Implement advanced techniques used in microbiology that offer significant advantages over traditional culture-based methods
Process Analytical Technology (PAT): Integrate real-time monitoring and control systems to enhance product quality and process understanding.
Data Analytics and Artificial Intelligence: Implement advanced data analysis tools to identify trends, predict maintenance needs, and optimize processes.
Conclusion
Maintaining a state-of-the-art biotech facility requires a proactive and systematic approach to technological upgrades. By establishing an ongoing process for identifying and implementing improvements, facilities can ensure compliance with FDA requirements, align with industry standards, and stay competitive in the rapidly evolving biotech landscape.
Remember that the goal is not just to meet current regulatory expectations but to anticipate future requirements and position your facility at the forefront of biotech manufacturing excellence. By following this comprehensive approach and staying informed on industry developments, you can create a robust, flexible, and compliant manufacturing environment that supports the production of high-quality biopharmaceutical products.
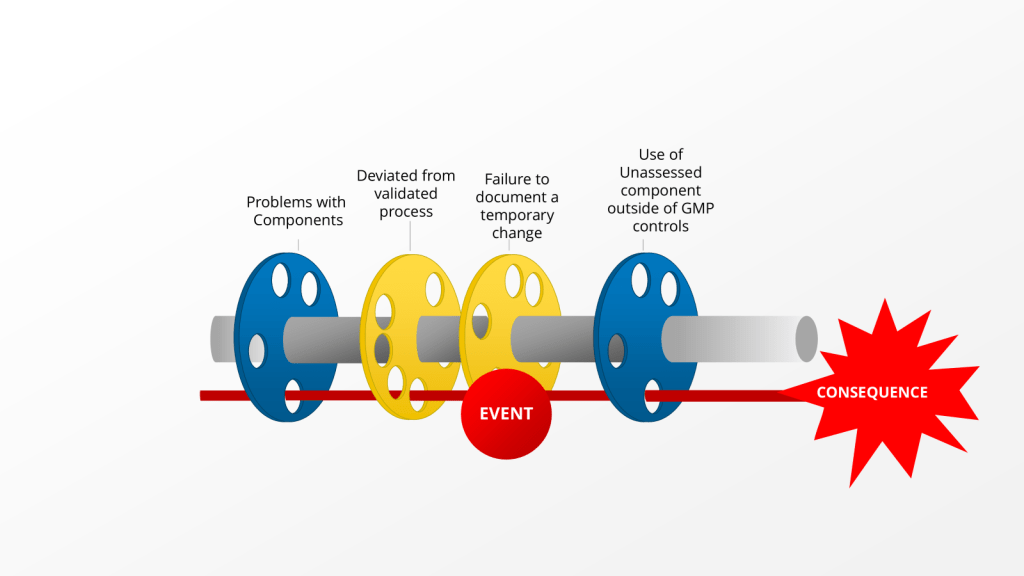
“there is no retrospective review of batch records for batches within expiry, to identify any other process deviations performed without the appropriate corresponding documentation including risk assessment(s).” – 2025 Warning Letter from the US FDA to Sanofi
This comment is about an instance where Sanofi deviated from the validated process by using an unvalidated single use component. Instead of self-identifying, creating a deviation and doing the right change control activities, the company just kept on deviating by using a non-controlled document.
This is a big problem for lots of reasons, from uncontrolled documents, to not using the change control system, to breaking the validated state. What the language quoted above really brings to bear is the question, when should we evaluate our records for other similar instances of this happening, so we can address it.
When a deviation investigation reveals recurring bad decision-making, it is crucial to expand the investigation and conduct a retrospective review of batch records. A good cutoff of this can be only for batches within expiry. This expanded investigation helps identify any other process deviations that may have occurred but were not discovered or documented at the time. Here’s when and how to approach this situation:
Triggers for Expanding the Investigation
Recurring Deviations: If the same or similar deviations are found to be recurring, it indicates a systemic issue that requires a broader investigation.
Pattern of Human Errors: When a pattern of human errors or poor decision-making is identified, it suggests potential underlying issues in training, procedures, or processes.
Critical Deviations: For deviations classified as critical, a more thorough investigation is typically warranted, including a retrospective review.
Potential Impact on Product Quality: If there’s a strong possibility that undiscovered deviations could affect product quality or patient safety, an expanded investigation becomes necessary.
Conducting the Retrospective Review
Timeframe: Review batch records for all batches within expiry, typically covering at least two years of production. Similarily for issues in the FUSE program you might look since the last requalification, or from a decide to go backwards in concentric circles based on what you find.
Scope: Examine not only the specific process where the deviation was found but also related processes or areas that could be affected. Reviewing related processes is critical.
Data Analysis: Utilize statistical tools and trending analysis techniques to identify patterns or anomalies in the historical data.
Cross-Functional Approach: Involve a team of subject matter experts from relevant departments to ensure a comprehensive review.
Documentation Review: Examine batch production records, laboratory control records, equipment logs, and any other relevant documentation.
Root Cause Analysis: Apply root cause analysis techniques to understand the underlying reasons for the recurring issues.
Key Considerations
Risk Assessment: Prioritize the review based on the potential risk to product quality and patient safety.
Data Integrity: Ensure that any retrospective data used is reliable and has maintained its integrity.
Corrective Actions: Develop and implement corrective and preventive actions (CAPAs) based on the findings of the expanded investigation.
Regulatory Reporting: Assess the need for notifying regulatory authorities based on the severity and impact of the findings.
By conducting a thorough retrospective review when recurring bad decision-making is identified, companies can uncover hidden issues, improve their quality systems, and prevent future deviations. This proactive approach not only enhances compliance but also contributes to continuous improvement in pharmaceutical manufacturing processes.
In the case of an issue that rises to a regulatory observation this becomes a firm must. The agency has raised a significant concern and they will want proof that this is a limited issue or that you are holistically dealing with it across the organization.
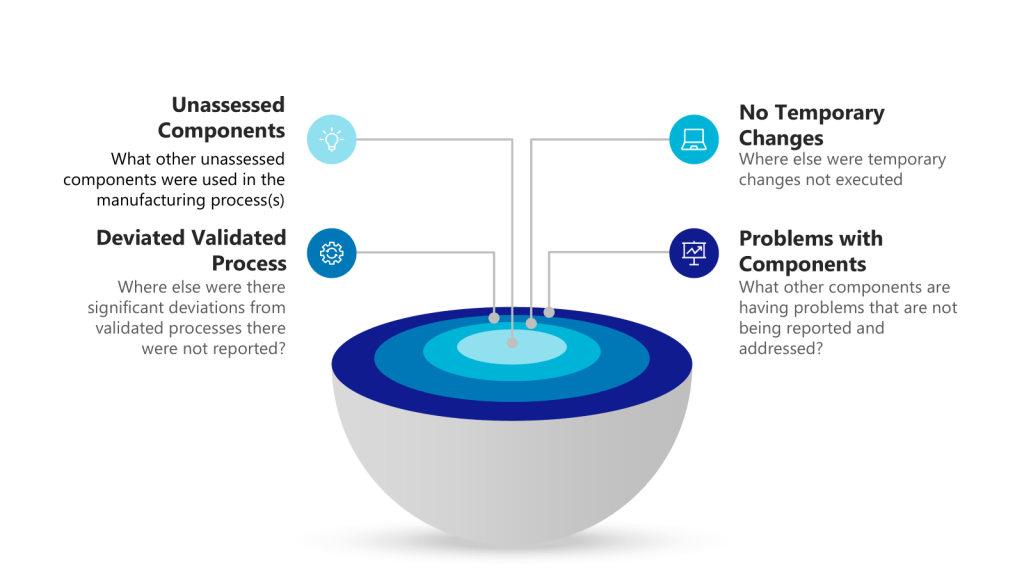
Concentric Circles of Investigation
Each layer of the investigation may require holistic looks. Utilizing the example above we have:
Layer of Problem
Further Investigationto Answer
Use of unassessed component outside of GMP controls
What other unassessed components were used in the manufacturing process(s)
In recent years, the importance of diversity in clinical trials has gained significant attention in the medical research community. This focus is not just a matter of inclusivity; it’s a crucial scientific and ethical imperative that directly impacts the quality and applicability of medical research.
Why Diversity in Clinical Trials is Essential
Scientific Validity and Generalizability
Different populations may respond differently to the same treatment due to variations in genetics, lifestyle, and environmental factors. By including diverse participants, researchers can better understand how a treatment works across various groups, leading to more accurate and widely applicable results.
Addressing Health Disparities
Minority groups often experience poorer health outcomes in various diseases. Including these groups in clinical trials is a crucial step towards understanding and addressing these disparities, potentially leading to more targeted and effective treatments for underserved populations.
Innovation and Discovery
Diversity in clinical trials can lead to unexpected discoveries. For instance, the identification of PCSK9, which revolutionized our understanding of cholesterol homeostasis, was a result of studying variations in cardiovascular risk factors among different racial groups.
Alignment with ICH Guidelines
The International Council for Harmonisation (ICH) has recognized the importance of diversity in its updated guidelines, particularly in ICH E6(R3) and ICH E8(R1).
ICH E6(R3)
This guideline emphasizes the importance of including diverse patient populations in clinical trials. It encourages the use of innovative trial designs and technologies to enable wider participation and inclusion of diverse populations. The guideline also stresses the need for quality by design (QbD) and a focus on critical-to-quality factors, which inherently includes considerations of diversity to ensure the reliability of trial results.
ICH E8(R1)
ICH E8(R1) focuses on the general considerations for clinical studies and emphasizes the importance of engaging with a broader range of stakeholders, including patients and patient advocacy groups. This approach naturally leads to more diverse perspectives in trial design and conduct, potentially increasing participation from underrepresented groups.
The Impact of Recent Policy Changes
The recent purge of FDA pages on clinical trial diversity, as reported by STAT News, raises significant concerns about the future of inclusive clinical. This action, part of a wider executive order banning diversity, equity, and inclusion (DEI) initiatives, could have far-reaching consequences:
Reduced Guidance: The removal of these resources may leave researchers and pharmaceutical companies with less clear direction on how to ensure diverse representation in their trials.
Potential Setbacks: Years of progress in improving trial diversity could be undermined, potentially leading to less representative studies and, consequently, less generalizable results.
Health Equity Concerns: This move could exacerbate existing health disparities by reducing the focus on including underrepresented groups in clinical research.
Scientific Integrity: The quality and applicability of clinical trial data may be compromised if diversity is not actively pursued, potentially affecting the safety and efficacy of new treatments for certain populations.
Moving Forward
Despite this setback, the scientific and pharma community must continue to prioritize diversity in clinical trials. The principles outlined in ICH E6(R3) and E8(R1) provide a strong foundation for this effort. Researchers, pharmaceutical companies, and regulatory bodies should:
Continue to develop innovative recruitment strategies to reach diverse populations.
Engage with community leaders and organizations to build trust and awareness about clinical trials.
Design trials with flexibility to improve access for all populations, including the use of decentralized trial elements.
Maintain a focus on quality by design, ensuring that diversity considerations are built into trial planning from the outset.
It is important to remember that E6(r3) is the regulation in Europe, while it is a guidance in the US. So companies need to follow it for their EMA approval possibilities.
In conclusion, diversity in clinical trials is not just a matter of equity; it’s a scientific necessity that ensures the development of safe and effective treatments for all populations. While recent policy changes may present challenges, the medical research community must remain committed to this crucial aspect of clinical research, guided by international standards and ethical imperatives.
In the previous post, we discussed the critical importance of thorough investigations into deviations, as highlighted by the recent FDA warning letter to Sanofi. Let us delve deeper into a specific aspect of these investigations: determining whether an invalidated out-of-specification (OOS) result for bioburden, endotoxin, or environmental monitoring action limit excursions conclusively demonstrates causative laboratory error.
When faced with an OOS result in microbiological testing, it’s crucial to conduct a thorough investigation before invalidating the result. The FDA expects companies to provide scientific justification and evidence that conclusively demonstrates a causative laboratory error if a result is to be invalidated.
Key Steps in Evaluating Laboratory Error
1. Review of Test Method and Procedure
Examine the standard operating procedure (SOP) for the test method
Verify that all steps were followed correctly
Check for any deviations from the established procedure
2. Evaluation of Equipment and Materials
Evaluation of Equipment and Materials is a critical step in determining whether laboratory error caused an out-of-specification (OOS) result, particularly for bioburden, endotoxin, or environmental monitoring tests. Here’s a detailed approach to performing this evaluation:
Run performance verification tests on key equipment used in the analysis
Review equipment logs for any recent malfunctions or irregularities
Verify that all equipment settings were correct for the specific test performed
Calibration Review
Check calibration records to ensure equipment was within its calibration period
Verify that calibration standards used were traceable and not expired
Review any recent calibration data for trends or shifts
Maintenance Evaluation
Examine maintenance logs for adherence to scheduled maintenance
Look for any recent repairs or adjustments that could affect performance
Verify that all preventive maintenance tasks were completed as required
Materials Evaluation
Reagent Quality Control
Check expiration dates of all reagents used in the test
Review storage conditions to ensure reagents were stored properly
Verify that quality control checks were performed on reagents before use
Media Assessment (for Bioburden and Environmental Monitoring)
Review growth promotion test results for culture media
Check pH and sterility of prepared media
Verify that media was stored at the correct temperature
Water Quality (for Endotoxin Testing)
Review records of water quality used for reagent preparation
Check for any recent changes in water purification systems
Verify endotoxin levels in water used for testing
Environmental Factors
Laboratory Conditions
Review temperature and humidity logs for the testing area
Check for any unusual events (e.g., power outages, HVAC issues) around the time of testing
Verify that environmental conditions met the requirements for the test method
Contamination Control
Examine cleaning logs for the laboratory area and equipment
Review recent environmental monitoring results for the testing area
Check for any breaches in aseptic technique during testing
Documentation Review
Standard Operating Procedures (SOPs)
Verify that the most current version of the SOP was used
Check for any recent changes to the SOP that might affect the test
Ensure all steps in the SOP were followed and documented
Equipment and Material Certifications
Review certificates of analysis for critical reagents and standards
Check equipment qualification documents (IQ/OQ/PQ) for compliance
Verify that all required certifications were current at the time of testing
By thoroughly evaluating equipment and materials using these detailed steps, laboratories can more conclusively determine whether an OOS result was due to laboratory error or represents a true product quality issue. This comprehensive approach helps ensure the integrity of microbiological testing and supports robust quality control in pharmaceutical manufacturing.
3. Assessment of Analyst Performance
Here are key aspects to consider when evaluating analyst performance during an OOS investigation:
Review Training Records
Examine the analyst’s training documentation to ensure they are qualified to perform the specific test method.
Verify that the analyst has completed all required periodic refresher training.
Check if the analyst has demonstrated proficiency in the particular test method recently.
Evaluate Recent Performance History
Review the analyst’s performance on similar tests over the past few months.
Look for any patterns or trends in the analyst’s results, such as consistently high or low readings.
Compare the analyst’s results with those of other analysts performing the same tests.
Assess any personal factors that could have affected the analyst’s performance, such as fatigue, illness, or personal stress.
Review the analyst’s work schedule leading up to the OOS result for any unusual patterns or extended hours.
By thoroughly assessing analyst performance using these methods, investigators can determine whether human error contributed to the OOS result and identify areas for improvement in training, procedures, or work environment. It’s important to approach this assessment objectively and supportively, focusing on systemic improvements rather than individual blame.
4. Examination of Environmental Factors
Review environmental monitoring data for the testing area
Check for any unusual events or conditions that could have affected the test
5. Data Analysis and Trending
Compare the OOS result with historical data and trends
Look for any patterns or anomalies that might explain the result
Conclusive vs. Inconclusive Evidence
Conclusive Evidence of Laboratory Error
To conclusively demonstrate laboratory error, you should be able to:
Identify a specific, documented error in the testing process
Reproduce the error and show how it leads to the OOS result
Demonstrate that correcting the error leads to an in-specification result
Examples of conclusive evidence might include:
Documented use of an expired reagent
Verified malfunction of testing equipment
Confirmed contamination of a negative control
Inconclusive Evidence
If the investigation reveals potential issues but cannot definitively link them to the OOS result, the evidence is considered inconclusive. This might include:
Minor deviations from SOPs that don’t clearly impact the result
Slight variations in environmental conditions
Analyst performance issues that aren’t directly tied to the specific test
Special Considerations for Microbiological Testing
Bioburden, endotoxin, and environmental monitoring tests present unique challenges due to their biological nature.
Bioburden Testing
Consider the possibility of sample contamination during collection or processing
Evaluate the recovery efficiency of the test method
Assess the potential for microbial growth during sample storage
Endotoxin Testing
Review the sample preparation process, including any dilution steps
Evaluate the potential for endotoxin masking or enhancement
Consider the impact of product formulation on the test method
Environmental Monitoring
Assess the sampling technique and equipment used
Consider the potential for transient environmental contamination
Evaluate the impact of recent cleaning or maintenance activities
Documenting the Investigation
Regardless of the outcome, it’s crucial to thoroughly document the investigation process. This documentation should include:
A clear description of the OOS result and initial observations
Detailed accounts of all investigative steps taken
Raw data and analytical results from the investigation
A comprehensive analysis of the evidence
A scientifically justified conclusion
Conclusion
Determining whether an invalidated OOS result conclusively demonstrates causative laboratory error requires a systematic, thorough, and well-documented investigation. For microbiological tests like bioburden, endotoxin, and environmental monitoring, this process can be particularly challenging due to the complex and sometimes variable nature of biological systems.
Remember, the goal is not to simply invalidate OOS results, but to understand the root cause and implement corrective and preventive actions. Only through rigorous investigation and continuous improvement can we ensure the quality and safety of pharmaceutical products. When investigating environmental and in-process results we are investigating the whole house of contamination control.