I was recently reviewing the updated Q9(R1) Annex 1- Q8/Q9/Q10 Questions & Answers (R5) related to ICH Q9(R1) Quality Risk Management (QRM) that were approved on 30 October 2024 and what they say about knowledge management. While there are some fun new questions asked, I particularly like “Do regulatory agencies expect to see a formal knowledge management approach during inspections?”
To which the answer was: “No. There is no regulatory requirement for a formal knowledge management system. However. it is expected that knowledge from different processes and systems is appropriately utilised. Note: ‘formal’ in this context means a structured approach using a recognised methodology or (IT-) tool, executing and documenting something in a transparent and detailed manner.”
What does appropriately utilized mean? What is the standard for determining it? The agencies are quite willing to leave that to you to figure out.
As usual I think it is valuable to agree upon a few core assumptions for what appropriate utilization of knowledge management might look like.
While rare, viral contamination events can have severe consequences, potentially impacting product quality, patient safety, and company reputation. And while a consent decree is a good way to grow your skills, I tend to prefer to avoid causing one to happen.
Luckily, regulatory bodies have provided comprehensive guidelines, with ICH Q5A(R2) being a cornerstone document. Let’s explore the best practices for viral risk management in biotech, drawing from ICH Q5A and other relevant guidances.
The Three Pillars of Viral Safety
ICH Q5A outlines three complementary approaches to control potential viral contamination:
Selection and testing of cell lines and raw materials
Assessment of viral clearance capacity in production processes
Testing of the product at appropriate stages for contaminating viruses
These pillars form the foundation of a robust viral safety strategy.
Cell Line and Raw Material Control
Thoroughly document the origin and history of cell lines
Implement comprehensive testing programs for cell banks, including master and working cell banks
Carefully assess and control animal-derived raw materials
Consider using chemically-defined or animal-free raw materials where possible
Implement stringent change control and quality agreements with raw material suppliers
Viral Clearance Capacity
Design manufacturing processes with multiple orthogonal viral clearance steps
Validate the effectiveness of viral clearance steps using model viruses
Aim for a cumulative viral reduction factor of at least 4 log10 per the USP guidelines
Consider both dedicated viral inactivation steps (e.g., low pH treatment) and removal steps (e.g., nanofiltration)
For continuous manufacturing, assess the impact of process dynamics on viral clearance
In-Process and Final Product Testing
Develop a comprehensive testing strategy for in-process materials and final product
Utilize state-of-the-art detection methods, including PCR and next-generation sequencing (NGS)
Consider replacing traditional in vivo assays with molecular methods where appropriate
Implement a testing program that covers a broad spectrum of potential viral contaminants
Risk-Based Approach
The revised ICH Q5A(R2) emphasizes a risk-based approach to viral safety. This involves:
Conducting thorough risk assessments of the entire manufacturing process
Identifying critical control points for viral contamination
Implementing appropriate mitigation strategies based on risk levels
Continuously monitoring and updating the risk assessment as new information becomes available
Prior knowledge, including “in-house” experience, plays a crucial role in viral risk assessment and management for biopharmaceutical manufacturing. Here’s how it can be effectively utilized:
Leveraging Historical Data
Review past viral contamination events or near-misses within the organization
Analyze trends in raw material quality and supplier performance
Evaluate the effectiveness of previous risk mitigation strategies
Process Design and Optimization
Apply lessons learned from previous manufacturing campaigns to improve process robustness
Use historical data to identify critical control points for viral contamination
Optimize viral clearance steps based on past validation studies
Cell Line Susceptibility
Use accumulated data on cell line susceptibility to various viruses to inform risk assessments
Apply knowledge of cell line behavior under different conditions to enhance contamination detection
Risk Assessment Approach
The risk assessment process should take a holistic approach, focusing on:
Raw material sourcing and testing
Identifying high-risk materials, especially animal-derived components
Assessing chemically-undefined components like hydrolysates and peptones
Evaluating materials produced or stored in non-controlled environments
Cell substrate selection and characterization
Documenting the derivation and source history of the cell line
Testing cell banks extensively for adventitious agents
Assessing the cell line’s susceptibility to various viruses
Process design for viral clearance
Designing manufacturing processes with multiple orthogonal viral clearance steps
Facility design and operations
Implementing robust cleaning and sanitization procedures
Ensuring proper facility layout and air handling systems to prevent contamination spread
Personnel training and practices
Training on proper gowning procedures and personal protective equipment (PPE) usage
Policies on illness reporting and exclusion of sick employees from critical areas
Preparedness and Response
While prevention is key, being prepared for a potential contamination event is crucial:
Develop a comprehensive viral contamination response plan[6]
Regularly practice and update the response plan through mock drills
Establish clear communication channels and decision-making processes
Prepare strategies for containment, decontamination, and facility restart
Continuous Improvement
Viral risk management is an ongoing process:
Stay updated on emerging technologies and regulatory guidance
Participate in industry forums and share best practices
Invest in employee training and awareness programs
Continuously evaluate and improve viral safety strategies
By implementing these best practices and adhering to regulatory guidances like ICH Q5A, we can strive to significantly mitigate the risk of viral contamination. While no approach can guarantee absolute safety, a comprehensive, risk-based strategy that leverages cutting-edge technologies and emphasizes preparedness will go a long way in protecting patients, products, and the industry as a whole.
In the last two posts (here and here) I’ve been talking about how process mapping is a valuable set of techniques to create a visual representation of the processes within an organization. Fundamental tools, every quality professional should be fluent in them.
The next level of maturity is process modeling which involves creating a digital representation of a process that can be analyzed, simulated, and optimized. Way more comprehensive, and frankly, very very hard to do and maintain.
Process Map
Process Model
Why is this Important?
Notation ambiguous
Standardized notation convention
Standardized notation conventions for process modeling, such as Business Process Model and Notation (BPMN), drive clarity, consistency, communication and process improvements.
Precision usually lacking
As precise as needed
Precision drives model accuracy and effectiveness. Too often process maps are all over the place.
Icons (representing process components made up or loosely defined
Icons are objectively defined and standardized
The use of common modeling conventions ensures that all process creators represent models consistently, regardless of who in the organization created them.
Relationship of icons portrayed visually
Icon relationships definite and explained in annotations, process model glossary, and process narratives
Reducing ambiguity, improving standardization and easing knowledge transfer are the whole goal here. And frankly, the average process map can fall really short.
Limited to portrayal of simple ideas
Can depict appropriate complexity
We need to strive to represent complex workflows in a visually comprehensible manner, striking a balance between detail and clarity. The ability to have scalable detail cannot be undersold.
One-time snapshot
Can grow, evolve, mature
How many times have you sat down to a project and started fresh with a process map? Enough said.
May be created with simple drawing tools
Created with a tool appropriate to the need
The right tool for the right job
Difficult to use for the simplest manual simulations
May provide manual or automated process simulation
In w world of more and more automation, being able to do a good process simulation is critical.
Difficult to link with related diagram or map
Vertical and horizontal linking, showing relationships among processes and different process levels
Processes don’t stand along, they are interconnected in a variety of ways. Being able to move up and down in detail and across the process family is great for diagnosing problems.
Uses simple file storage with no inherent relationships
Uses a repository of related models within a BPM system
It is fairly common to do process maps and keep them separate, maybe in an SOP, but more often in a dozen different, unconnected places, making it difficult to put your hands on it. Process modeling maturity moves us towards a library approach, with drives knowledge management.
Appropriate for quick capture of ideas
Appropriate for any level of process capture, analysis and design
Processes are living and breathing, our tools should take that into account.
This is all about moving to a process repository and away from a document mindset. I think it is a great shame that the eQMS players don’t consider this part of their core mission. This is because most quality units don’t see this as part of their core mission. We as quality leaders should be seeing process management as critical for future success. This is all about profound knowledge and utilizing it to drive true improvements.
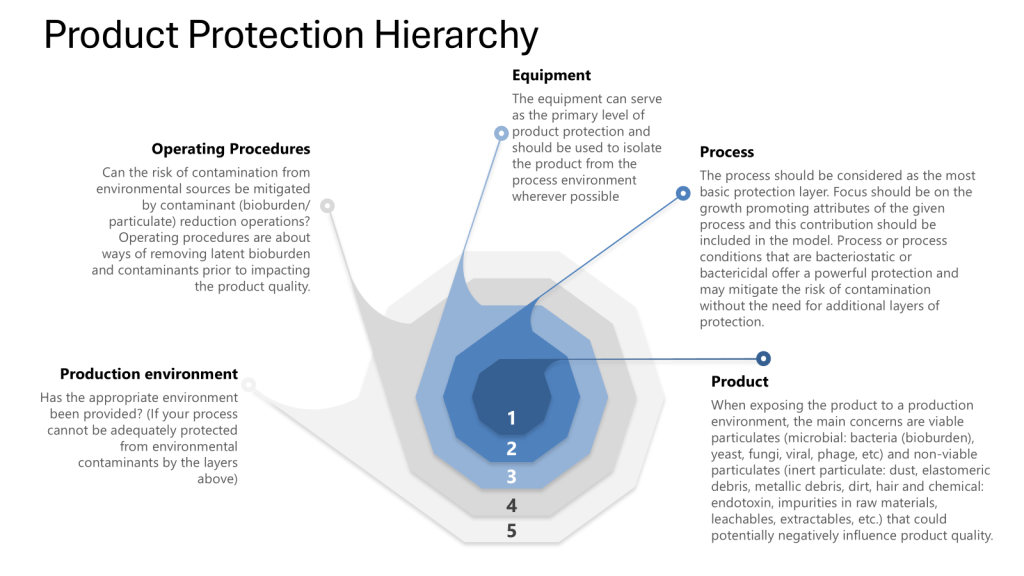
Maintaining process closure is crucial for ensuring product quality and safety in biotechnology manufacturing, especially when using single-use systems (SUS). This approach is an integral part of the contamination control strategy (CCS). To validate process closure in SUS-based biotech manufacturing, a comprehensive method is necessary, incorporating:
Risk assessment
Thorough testing
Ongoing monitoring
By employing risk analysis tools such as Hazard Analysis and Critical Control Points (HACCP) and Failure Mode and Effects Analysis (FMEA), manufacturers can identify potential weaknesses in their processes. Additionally, addressing all four layers of protection helps ensure process integrity and product safety. This risk-based approach to process closure validation is essential for maintaining the high standards required in biotechnology manufacturing, including meeting Annex 1.
Understanding Process Closure
Process closure refers to the isolation of the manufacturing process from the external environment to prevent contamination. In biotech, this is particularly crucial due to the sensitivity of biological products and the potential for microbial contamination.
Throughout this process it is important to apply the four layers of protection that form the foundation of a robust contamination control strategy:
Process: The inherent ability of the process to prevent or control contamination
Equipment: The design and functionality of equipment to maintain closure
Operating Procedures: The practices and protocols followed by personnel
Production Environment: The controlled environment surrounding the process
I was discussing this with some colleagues this week (preparing for some risk assessments) and I was reminded that we really should put the Patient in at the center, the zero. Truer words have never been spoken as the patient truly is our zeroth law, the fundamental principle of the GxPs.
Key Steps for Validating Process Closure
Risk Assessment
Start with a comprehensive risk assessment using tools such as HACCP (Hazard Analysis and Critical Control Points) and FMEA (Failure Mode and Effects Analysis). It is important to remember this is not a one or another, but a multi-tiered approach where you first determine the hazards through the HACCP and then drill down into failures through an FMEA.
HACCP Approach
In the HACCP we will apply a systematic, preventative approach to identify hazards in the process with the aim to produce a documented plan to control these scenarios.
a) Conduct a hazard analysis b) Identify Critical Control Points (CCPs) c) Establish critical limits d) Implement monitoring procedures e) Define corrective actions f) Establish verification procedures g) Maintain documentation and records
FMEA Considerations
In the FMEA we will look for ways the process fails, focusing on the SUS components. We will evaluate failures at each level of control (process, equipment, operating procedure and environment).
Identify potential failure modes in the SUS components
Assess the severity, occurrence, and detectability of each failure mode
Calculate Risk Priority Numbers (RPN) to prioritize risks
Verification
Utilizing these risk assessments, define the user requirements specification (URS) for the SUS, focusing on critical aspects that could impact product quality and patient safety. This should include:
Process requirements (e.g. working volumes, flow rates, pressure ranges)
Following the ASTM E2500 approach, when we conduct the design review of the proposed SUS configuration, to evaluate how well it meets the URS, we want to ensure we cover:
Overall system design and component selection
Materials of construction
Sterilization/sanitization approach
Integrity assurance measures
Sampling and monitoring capabilities
Automation and control strategy
Circle back to the HACCP and FMEA to ensure they appropriately cover critical aspects like:
Loss of sterility/integrity
Leachables/extractables introduction
Bioburden control failures
Cross-contamination risks
Process parameter deviations
These risk assessments will define critical control parameters and acceptance criteria based on the risk assessment. These will form the basis for verification testing. We will through our verification plan have an appropriate approach to:
Verify proper installation of SUS components
Check integrity of connections and seals
Confirm correct placement of sensors and monitoring devices
Document as-built system configuration
Test system integrity under various operating conditions
Perform leak tests on connections and seals
Validate sterilization processes for SUS components
Verify functionality of critical sensors and control
Run simulated production cycles
Monitor for contamination using sensitive detection methods
Verify maintenance of sterility throughout the process
Assess product quality attributes
The verification strategy will leverage a variety of supplier documentation and internal testing.
Closure Analysis Risk Assessment (CLARA)
Acceptance and release will be to perform a detailed CLARA to:
Identify all potential points of contamination ingress
Assess the effectiveness of closure mechanisms
Evaluate the robustness of aseptic connections
Determine the impact of manual interventions on system closure
On Going Use
Coming out of our HACCP we will have a monitoring and verification plan, this will include some important aspects based on our CCPs.
Integrity Testing
Implement routine integrity testing protocols for SUS components
Utilize methods such as pressure decay tests or helium leak detection
Establish acceptance criteria for integrity tests
Environmental Monitoring
Develop a comprehensive environmental monitoring program
Include viable and non-viable particle monitoring
Establish alert and action limits for environmental contaminants
Establish a robust change control process for any modifications to the SUS or process
Regularly review and update risk assessments based on new data or changes
Implement a continuous improvement program to enhance process closure
Leveraging the Four Layers of Protection
Throughout the validation process, ensure that each layer of protection is addressed:
Process:
Optimize process parameters to minimize contamination risks
Implement in-process controls to detect deviations
Equipment:
Validate the design and functionality of SUS components
Ensure proper integration of SUS with existing equipment
Operating Procedures:
Develop and validate aseptic techniques for SUS handling
Implement procedures for system assembly and disassembly
Production Environment:
Qualify the cleanroom environment
Validate HVAC systems and air filtration
Remember that validation is an ongoing process. Regular reviews, updates to risk assessments, and incorporation of new technologies and best practices are essential for maintaining a state of control in biotech manufacturing using single-use systems.
Connected to the Contamination Control Strategy
Closed systems are a key element of the overall contamination control strategy with closed processing and closed systems now accepted as the most effective contamination control risk mitigation strategy. I might not be able to manufacture in the woods yet, but darn if I won’t keep trying.
They serve as a primary barrier to prevent contamination from the manufacturing environment by helping to mitigate the risk of contamination by isolating the product from the surrounding environment. Closed systems are the key protective measure to prevent contamination from the manufacturing environment and cross-contamination from neighboring operations.
The risk assessments leveraged during the implementation of closed systems are a crucial part of developing an effective CCS and will communicate the (ideally) robust methods used to protect products from environmental contamination and cross-contamination. This is tied into the facility design, environmental controls, risk assessments, and overall manufacturing strategies, which are the key components of a comprehensive CCS.
The key is to use a systematic, science-based approach to identify potential hazards at each layer and implement appropriate preventive controls. The controls should be validated, monitored, verified and documented as part of the overall contamination control strategy (system). Regular review and updates are needed to ensure the controls remain effective.