Maturity models offer significant benefits to organizations by providing a structured framework for benchmarking and assessment. Organizations can clearly understand their strengths and weaknesses by evaluating their current performance and maturity level in specific areas or processes. This assessment helps identify areas for improvement and sets a baseline for measuring progress over time. Benchmarking against industry standards or best practices also allows organizations to see how they compare to their peers, fostering a competitive edge.
One of the primary advantages of maturity models is their role in fostering a culture of continuous improvement. They provide a roadmap for growth and development, encouraging organizations to strive for higher maturity levels. This continuous improvement mindset helps organizations stay agile and adaptable in a rapidly changing business environment. By setting clear goals and milestones, maturity models guide organizations in systematically addressing deficiencies and enhancing their capabilities.
Standardization and consistency are also key benefits of maturity models. They help establish standardized practices across teams and departments, ensuring that processes are executed with the same level of quality and precision. This standardization reduces variability and errors, leading to more reliable and predictable outcomes. Maturity models create a common language and framework for communication, fostering collaboration and alignment toward shared organizational goals.
The use of maturity models significantly enhances efficiency and effectiveness. Organizations can increase productivity and use their resources by identifying areas for streamlining operations and optimizing workflows. This leads to reduced errors, minimized rework, and improved process efficiency. The focus on continuous improvement also means that organizations are constantly seeking ways to refine and enhance their operations, leading to sustained gains in efficiency.
Maturity models play a crucial role in risk reduction and compliance. They assist organizations in identifying potential risks and implementing measures to mitigate them, ensuring compliance with relevant regulations and standards. This proactive approach to risk management helps organizations avoid costly penalties and reputational damage. Moreover, maturity models improve strategic planning and decision-making by providing a data-backed foundation for setting priorities and making informed choices.
Finally, maturity models improve communication and transparency within organizations. Providing a common communication framework increases transparency and builds trust among employees. This improved communication fosters a sense of shared purpose and collaboration, essential for achieving organizational goals. Overall, maturity models serve as valuable tools for driving continuous improvement, enhancing efficiency, and fostering a culture of excellence within organizations.
Business Process Maturity Model (BPMM)
A structured framework used to assess and improve the maturity of an organization’s business processes, it provides a systematic methodology to evaluate the effectiveness, efficiency, and adaptability of processes within an organization, guiding continuous improvement efforts.
Key Characteristics of BPMM
Assessment and Classification: BPMM helps organizations understand their current process maturity level and identify areas for improvement. It classifies processes into different maturity levels, each representing a progressive improvement in process management.
Guiding Principles: The model emphasizes a process-centric approach focusing on continuous improvement. Key principles include aligning improvements with business goals, standardization, measurement, stakeholder involvement, documentation, training, technology enablement, and governance.
Incremental Levels
BPMM typically consists of five levels, each building on the previous one:
- Initial: Processes are ad hoc and chaotic, with little control or consistency.
- Managed: Basic processes are established and documented, but results may vary.
- Standardized: Processes are well-documented, standardized, and consistently executed across the organization.
- Predictable: Processes are quantitatively measured and controlled, with data-driven decision-making.
- Optimizing: Continuous process improvement is ingrained in the organization’s culture, focusing on innovation and optimization.
Benefits of BPMM
- Improved Process Efficiency: By standardizing and optimizing processes, organizations can achieve higher efficiency and consistency, leading to better resource utilization and reduced errors.
- Enhanced Customer Satisfaction: Mature processes lead to higher product and service quality, which improves customer satisfaction.
- Better Change Management: Higher process maturity increases an organization’s ability to navigate change and realize project benefits.
- Readiness for Technology Deployment: BPMM helps ensure organizational readiness for new technology implementations, reducing the risk of failure.
Usage and Implementation
- Assessment: Organizations can conduct BPMM assessments internally or with the help of external appraisers. These assessments involve reviewing process documentation, interviewing employees, and analyzing process outputs to determine maturity levels.
- Roadmap for Improvement: Organizations can develop a roadmap for progressing to higher maturity levels based on the assessment results. This roadmap includes specific actions to address identified deficiencies and improve process capabilities.
- Continuous monitoring and regular evaluations are crucial to ensure that processes remain effective and improvements are sustained over time.
A BPMM Example: Validation Program based on ASTM E2500
To apply the Business Process Maturity Model (BPMM) to a validation program aligned with ASTM E2500, we need to evaluate the program’s maturity across the five levels of BPMM while incorporating the key principles of ASTM E2500. Here’s how this application might look:
Level 1: Initial
At this level, the validation program is ad hoc and lacks standardization:
- Validation activities are performed inconsistently across different projects or departments.
- There’s limited understanding of ASTM E2500 principles.
- Risk assessment and scientific rationale for validation activities are not systematically applied.
- Documentation is inconsistent and often incomplete.
Level 2: Managed
The validation program shows some structure but lacks organization-wide consistency:
- Basic validation processes are established but may not fully align with ASTM E2500 guidelines.
- Some risk assessment tools are used, but not consistently across all projects.
- Subject Matter Experts (SMEs) are involved, but their roles are unclear.
- There’s increased awareness of the need for scientific justification in validation activities.
Level 3: Standardized
The validation program is well-defined and consistently implemented:
- Validation processes are standardized across the organization and align with ASTM E2500 principles.
- Risk-based approaches are consistently used to determine the scope and extent of validation activities.
- SMEs are systematically involved in the design review and verification processes.
- The concept of “verification” replaces traditional IQ/OQ/PQ, focusing on critical aspects that impact product quality and patient safety.
- Quality risk management tools (e.g., impact assessments, risk management) are routinely used to identify critical quality attributes and process parameters.
Level 4: Predictable
The validation program is quantitatively managed and controlled:
- Key Performance Indicators (KPIs) for validation activities are established and regularly monitored.
- Data-driven decision-making is used to continually improve the efficiency and effectiveness of validation processes.
- Advanced risk management techniques are employed to predict and mitigate potential issues before they occur.
- There’s a strong focus on leveraging supplier documentation and expertise to streamline validation efforts.
- Engineering procedures for quality activities (e.g., vendor technical assessments and installation verification) are formalized and consistently applied.
Level 5: Optimizing
The validation program is characterized by continuous improvement and innovation:
- There’s a culture of continuous improvement in validation processes, aligned with the latest industry best practices and regulatory expectations.
- Innovation in validation approaches is encouraged, always maintaining alignment with ASTM E2500 principles.
- The organization actively contributes to developing industry standards and best practices in validation.
- Validation activities are seamless integrated with other quality management systems, supporting a holistic approach to product quality and patient safety.
- Advanced technologies (e.g., artificial intelligence, machine learning) may be leveraged to enhance risk assessment and validation strategies.
Key Considerations for Implementation
- Risk-Based Approach: At higher maturity levels, the validation program should fully embrace the risk-based approach advocated by ASTM E2500, focusing efforts on aspects critical to product quality and patient safety.
- Scientific Rationale: As maturity increases, there should be a stronger emphasis on scientific understanding and justification for validation activities, moving away from a checklist-based approach.
- SME Involvement: Higher maturity levels should see increased and earlier involvement of SMEs in the validation process, from equipment selection to verification.
- Supplier Integration: More mature programs will leverage supplier expertise and documentation effectively, reducing redundant testing and improving efficiency.
- Continuous Improvement: At the highest maturity level, the validation program should have mechanisms in place for continuous evaluation and improvement of processes, always aligned with ASTM E2500 principles and the latest regulatory expectations.
Process and Enterprise Maturity Model (PEMM),
The Process and Enterprise Maturity Model (PEMM), developed by Dr. Michael Hammer, is a comprehensive framework designed to help organizations assess and improve their process maturity. It is a corporate roadmap and benchmarking tool for companies aiming to become process-centric enterprises.
Key Components of PEMM
PEMM is structured around two main dimensions: Process Enablers and Organizational Capabilities. Each dimension is evaluated on a scale to determine the maturity level.
Process Enablers
These elements directly impact the performance and effectiveness of individual processes. They include:
- Design: The structure and documentation of the process.
- Performers: The individuals or teams executing the process.
- Owner: The person responsible for the process.
- Infrastructure: The tools, systems, and resources supporting the process.
- Metrics: The measurements used to evaluate process performance.
Organizational Capabilities
These capabilities create an environment that supports and sustains high-performance processes. They include:
- Leadership: The commitment and support from top management.
- Culture: The organizational values and behaviors that promote process excellence.
- Expertise: The skills and knowledge required to manage and improve processes.
- Governance: The mechanisms to oversee and guide process management activities.
Maturity Levels
Both Process Enablers and Organizational Capabilities are assessed on a scale from P0 to P4 (for processes) and E0 to E4 (for enterprise capabilities):
- P0/E0: Non-existent or ad hoc processes and capabilities.
- P1/E1: Basic, but inconsistent and poorly documented.
- P2/E2: Defined and documented, but not fully integrated.
- P3/E3: Managed and measured, with consistent performance.
- P4/E4: Optimized and continuously improved.
Benefits of PEMM
- Self-Assessment: PEMM is designed to be simple enough for organizations to conduct their own assessments without needing external consultants.
- Empirical Evidence: It encourages the collection of data to support process improvements rather than relying on intuition.
- Engagement: Involves all levels of the organization in the process journey, turning employees into advocates for change.
- Roadmap for Improvement: Provides a clear path for organizations to follow in their process improvement efforts.
Application of PEMM
PEMM can be applied to any type of process within an organization, whether customer-facing or internal, core or support, transactional or knowledge-intensive. It helps organizations:
- Assess Current Maturity: Identify the current state of process and enterprise capabilities.
- Benchmark: Compare against industry standards and best practices.
- Identify Improvements: Pinpoint areas that need enhancement.
- Track Progress: Monitor the implementation and effectiveness of process improvements.
A PEMM Example: Validation Program based on ASTM E2500
To apply the Process and Enterprise Maturity Model (PEMM) to an ASTM E2500 validation program, we can evaluate the program’s maturity across the five process enablers and four enterprise capabilities defined in PEMM. Here’s how this application might look:
Process Enablers
Design:
- P-1: Basic ASTM E2500 approach implemented, but not consistently across all projects
- P-2: ASTM E2500 principles applied consistently, with clear definition of requirements, specifications, and verification activities
- P-3: Risk-based approach fully integrated into design process, with SME involvement from the start
- P-4: Continuous improvement of ASTM E2500 implementation based on lessons learned and industry best practices
Performers:
- P-1: Some staff trained on ASTM E2500 principles
- P-2: All relevant staff trained and understand their roles in the ASTM E2500 process
- P-3: Staff proactively apply risk-based thinking and scientific rationale in validation activities
- P-4: Staff contribute to improving the ASTM E2500 process and mentor others
- P-1: Validation program has a designated owner, but role is not well-defined
- P-2: Clear ownership of the ASTM E2500 process with defined responsibilities
- P-3: Owner actively manages and improves the ASTM E2500 process
- P-4: Owner collaborates across departments to optimize the validation program
Infrastructure:
- P-1: Basic tools in place to support ASTM E2500 activities
- P-2: Integrated systems for managing requirements, risk assessments, and verification activities
- P-3: Advanced tools for risk management and data analysis to support decision-making
- P-4: Cutting-edge technology leveraged to enhance efficiency and effectiveness of the validation program
Metrics:
- P-1: Basic metrics tracked for validation activities
- P-2: Comprehensive set of metrics established to measure ASTM E2500 process performance
- P-3: Metrics used to drive continuous improvement of the validation program
- P-4: Predictive analytics used to anticipate and prevent issues in validation activities
Enterprise Capabilities
Leadership:
- E-1: Leadership aware of ASTM E2500 principles
- E-2: Leadership actively supports ASTM E2500 implementation
- E-3: Leadership drives cultural change to fully embrace risk-based validation approach
- E-4: Leadership promotes ASTM E2500 principles beyond the organization, influencing industry standards
Culture:
- E-1: Some recognition of the importance of risk-based validation
- E-2: Culture of quality and risk-awareness developing across the organization
- E-3: Strong culture of scientific thinking and continuous improvement in validation activities
- E-4: Innovation in validation approaches encouraged and rewarded
Expertise:
- E-1: Basic understanding of ASTM E2500 principles among key staff
- E-2: Dedicated team of ASTM E2500 experts established
- E-3: Deep expertise in risk-based validation approaches across multiple departments
- E-4: Organization recognized as thought leader in ASTM E2500 implementation
Governance:
- E-1: Basic governance structure for validation activities in place
- E-2: Clear governance model aligning ASTM E2500 with overall quality management system
- E-3: Cross-functional governance ensuring consistent application of ASTM E2500 principles
- E-4: Governance model that adapts to changing regulatory landscape and emerging best practices
To use this PEMM assessment:
- Evaluate your validation program against each enabler and capability, determining the current maturity level (P-1 to P-4 for process enablers, E-1 to E-4 for enterprise capabilities).
- Identify areas for improvement based on gaps between current and desired maturity levels.
- Develop action plans to address these gaps, focusing on moving to the next maturity level for each enabler and capability.
- Regularly reassess the program to track progress and adjust improvement efforts as needed.
Comparison Table
| Aspect | BPMM | PEMM |
|---|---|---|
| Creator | Object Management Group (OMG) | Dr. Michael Hammer |
| Purpose | Assess and improve business process maturity | Roadmap and benchmarking for process-centricity |
| Structure | Five levels: Initial, Managed, Standardized, Predictable, Optimizing | Two components: Process Enablers (P0-P4), Organizational Capabilities (E0-E4) |
| Focus | Process-centric, incremental improvement | Process enablers and organizational capabilities |
| Assessment Method | Often requires external appraisers | Designed for self-assessment |
| Guiding Principles | Standardization, measurement, continuous improvement | Empirical evidence, simplicity, organizational engagement |
| Applications | Enterprise systems, business process improvement, benchmarking | Process reengineering, organizational engagement, benchmarking |
In summary, while both BPMM and PEMM aim to improve business processes, BPMM is more structured and detailed, often requiring external appraisers, and focuses on incremental process improvement across organizational boundaries. In contrast, PEMM is designed for simplicity and self-assessment, emphasizing the role of process enablers and organizational capabilities to foster a supportive environment for process improvement. Both have advantages, and keeping both in mind while developing processes is key.



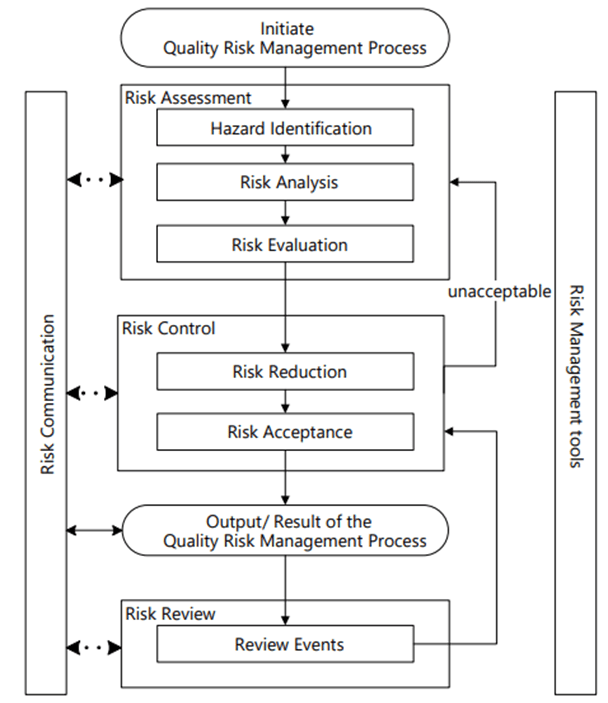

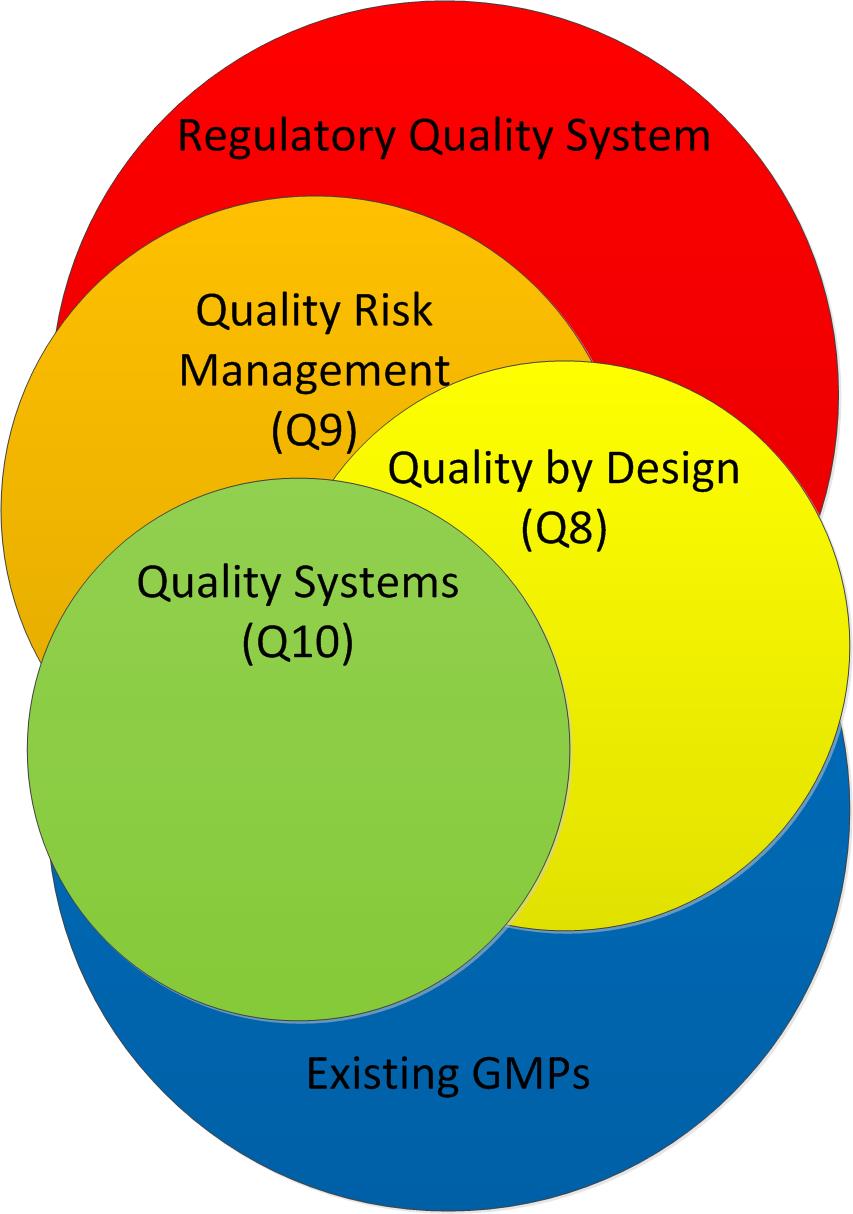{kind=link}