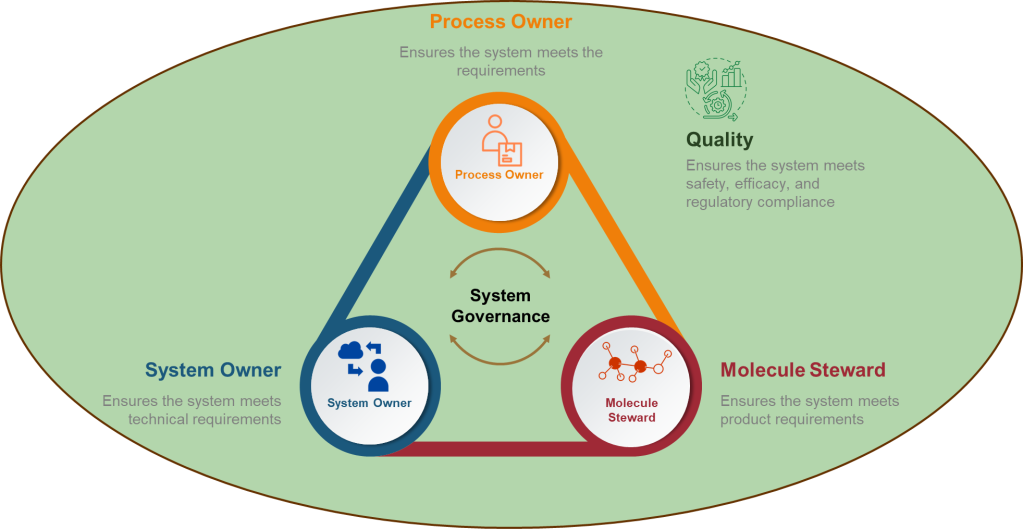
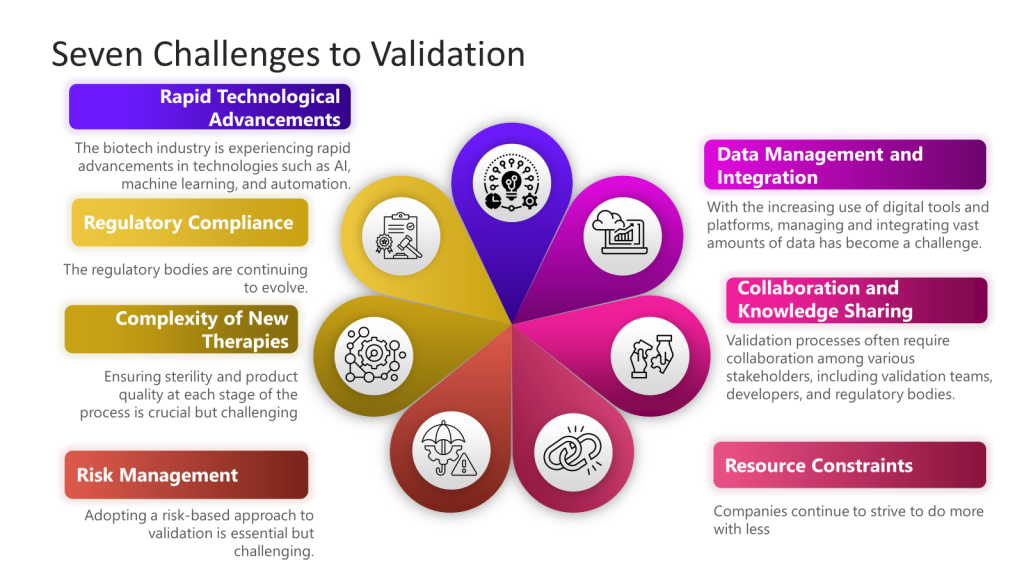
The terms “method qualification” and “method validation” are often used in the context of analytical procedures in the pharmaceutical and biotechnology industries. While related, they serve different purposes and are applied at different stages of method development. Here is a detailed comparison of the two:
Method Qualification
Definition
Method qualification demonstrates that an analytical method is suitable for its intended use during the early stages of development. It involves preliminary testing to ensure the method can produce reliable and reproducible results for the specific application.
Purpose
- Early Development: This is typically performed during the early phases of drug development (e.g., preclinical and Phase I clinical trials) to assess the method’s feasibility.
- Optimization: Helps in optimizing the method to ensure it meets the necessary performance criteria before full validation.
- Feasibility Studies: Often referred to as feasibility or pre-validation studies, method qualification helps understand the method’s performance characteristics and establish preliminary acceptance criteria.
Characteristics
- Flexibility: The method can still be modified and optimized based on the results obtained during qualification.
- Parameters: This evaluation method evaluates fewer parameters than validation, focusing on key performance indicators like specificity, linearity, accuracy, and precision.
- Voluntary: This is not always required by regulatory authorities, but it is a good practice to ensure the method is on the right track for future validation.
Example
A company developing a new drug might perform method qualification to ensure that their analytical method can accurately measure the drug’s concentration in biological samples before moving on to more rigorous validation studies.
Method Validation
Definition
Method validation proves that an analytical method is suitable for its intended purpose and can consistently produce reliable and reproducible results under specified conditions. It is a regulatory requirement for methods used to test drug substances and products.
Purpose
- Regulatory Compliance: Required by regulatory authorities (e.g., FDA, EMA) for methods used in quality control and release testing of pharmaceutical products.
- Late Development: This is typically performed during the later stages of drug development (e.g., Phase III clinical trials, and commercial production) when the method is fully developed and optimized.
- Consistency: Ensures that the method produces consistent results over time and across different laboratories.
Characteristics
- Rigidity: The method must be fully developed and optimized before validation. Any changes to the method after validation would require re-validation.
- Comprehensive: Involves a thorough evaluation of multiple parameters as defined by guidelines such as ICH Q2(R1), including accuracy, precision, specificity, linearity, range, limit of detection (LOD), limit of quantification (LOQ), robustness, and ruggedness.
- Mandatory: Required by regulatory authorities to ensure the quality, reliability, and consistency of analytical results.
Example
Before a pharmaceutical company can market a new drug, it must validate its analytical methods to demonstrate that they can reliably measure the drug’s potency, purity, and stability according to regulatory standards.
Key Differences
| Aspect | Method Qualification | Method Validation |
|---|---|---|
| Stage of Development | Early stages (pre-clinical, Phase I) | Later stages (Phase III, commercial production) |
| Purpose | Feasibility and optimization | Regulatory compliance and consistency |
| Flexibility | Method can be modified | Method must be fully developed and optimized |
| Parameters Evaluated | Fewer, key performance indicators | Comprehensive, as per regulatory guidelines |
| Regulatory Requirement | Voluntary, good practice | Mandatory |
In summary, method qualification is an early-stage activity aimed at ensuring that an analytical method is on the right path to becoming reliable and reproducible, while method validation is a more rigorous, comprehensive process required to demonstrate that the method meets all regulatory requirements for its intended use.