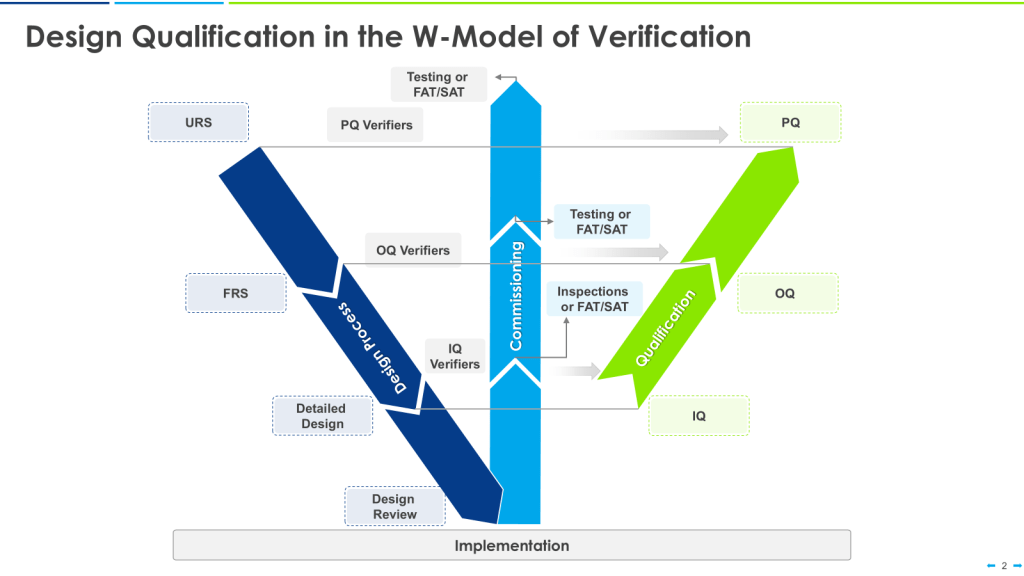
A critical step in ensuring the quality and safety of processes as part of verification is Design Review, which is sometimes expanded to Design Qualification.
Design Review is a systematic, documented examination of a proposed design to evaluate its adequacy and identify potential issues early in the development process. Here’s how to conduct an effective Design Review:
Plan Systematically: Schedule reviews at appropriate stages of development, ensuring they align with your project timeline.
Involve the Right People: Include representatives from all relevant functions and an independent reviewer not directly responsible for the design stage being evaluated.
Focus on Critical Aspects: Prioritize design elements that directly impact product quality and patient safety.
Document Thoroughly: Record all findings, including the design under review, participants, date, and any proposed actions.
Iterate as Needed: Conduct reviews iteratively as supplier design documents are published, allowing for early issue identification and correction.
Design Qualification: Verifying Suitability
Design Qualification (DQ) is the documented verification that the proposed design of facilities, equipment, or systems is suitable for its intended purpose. Here’s how to implement DQ effectively:
Develop User Requirements: Create a detailed User Requirements Specification (URS) outlining what the equipment or system is expected to do.
Create Functional Specifications: Translate user requirements into technical specifications that guide the design process.
Perform Risk Assessment: Identify potential risks associated with the design and develop mitigation strategies.
Review Design Specifications: Ensure the design meets all specified requirements, including GMP and regulatory standards.
Document and Approve: Formally document the DQ process and obtain approval from key stakeholders, including quality assurance personnel.
Integrating Design Review and DQ
To maximize the effectiveness of these processes:
Use a Risk-Based Approach: Prioritize efforts based on the level of risk to product quality and patient safety.
Leverage Subject Matter Experts: Involve SMEs from the start to contribute their expertise throughout the process.
Implement Change Management: Establish a robust system to manage design changes effectively and avoid late-stage issues.
Ensure Quality Oversight: Have Quality Assurance provide oversight to maintain compliance with current regulations and GMP requirements.
Document Comprehensively: Maintain thorough records of all reviews, qualifications, and decisions made during the process.
Implementing a systematic approach to Design Review and Design Qualification not only helps meet regulatory expectations but also contributes to operational efficiency and product excellence. As the pharmaceutical landscape evolves, staying committed to these foundational practices will remain crucial for success in this highly regulated industry.
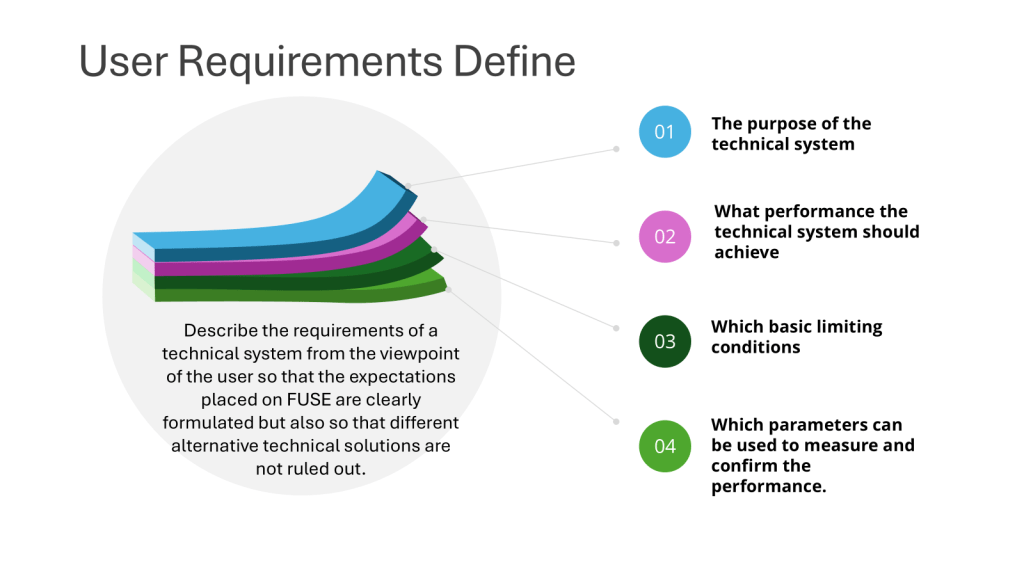
User requirements are typically divided into several categories to ensure comprehensive coverage of all aspects of product development, manufacturing, and quality control and to help guide the risk-based approach to verification.
Quality requirements focus on ensuring that the product meets all necessary quality standards and regulatory compliance. This category includes:
Good Manufacturing Practices (GMP) compliance, including around cleaning, cross-contamination, etc to ensure compliance with various regulations such as FDA guidelines, EU GMP, and ICH standards.
Documentation and record-keeping standards
Contamination control strategies are a key part of quality requirements, as they are essential for maintaining product quality and patient safety.
Data integrity requirements fall under this category, as they are crucial for ensuring the quality and reliability of data.
Not everyone advocates for this breakdown but I am a huge proponent as it divides the product specific requirements for the more standard must’s of meeting the cGMPs that are not product specific. This really helps when you are a multi-product facility and it helps define what is in the PQ versus what is in the PPQ.
Safety User Requirements
Safety requirements address the safety of personnel, patients, and the environment. They encompass:
Occupational health and safety measures
Environmental protection protocols
Patient safety considerations in product design
General User Requirements
General requirements cover broader aspects of the manufacturing system and facility. These may include:
Facility design and layout
Equipment specifications
Utility requirements (e.g., power, water, HVAC)
Maintenance procedures
By categorizing user requirements in this way, pharmaceutical companies can ensure a comprehensive approach to product development and manufacturing, addressing all critical aspects from product quality to regulatory compliance and safety. This will help drive appropriate verification.
“The specification for equipment, facilities, utilities or systems should be defined in a URS and/or a functional specification. The essential elements of quality need to be built in at this stage and any GMP risks mitigated to an acceptable level. The URS should be a point of reference throughout the validation life cycle.” – Annex 15, Section 3.2, Eudralex Volume 4
User Requirement Specifications serve as a cornerstone of quality in pharmaceutical manufacturing. They are not merely bureaucratic documents but vital tools that ensure the safety, efficacy, and quality of pharmaceutical products.
Defining the Essentials
A well-crafted URS outlines the critical requirements for facilities, equipment, utilities, systems and processes in a regulated environment. It captures the fundamental aspects and scope of users’ needs, ensuring that all stakeholders have a clear understanding of what is expected from the final product or system.
The phrase “essential elements of quality need to be built in at this stage” emphasizes the proactive approach to quality assurance. By incorporating quality considerations from the outset, manufacturers can:
Reduce the need for costly corrections later in the process
Ensure compliance with Good Manufacturing Practice (GMP) standards
Mitigating GMP Risks
Risk management is a crucial aspect of pharmaceutical manufacturing. The URS plays a vital role in identifying and addressing potential GMP risks early in the development process. By doing so, manufacturers can:
Ensure that the final product meets regulatory requirements
The URS as a Living Document
One of the key points in the regulations is that the URS should be “a point of reference throughout the validation life cycle.” This underscores the dynamic nature of the URS and its ongoing importance.
Continuous Reference
Throughout the development, implementation, and operation of a system or equipment, the URS serves as:
A benchmark for assessing progress
A guide for making decisions
A tool for resolving disputes or clarifying requirements
Adapting to Change
As projects evolve, the URS may need to be updated to reflect new insights, technological advancements, or changing regulatory requirements. This flexibility ensures that the final product remains aligned with user needs and regulatory expectations.
Practical Implications
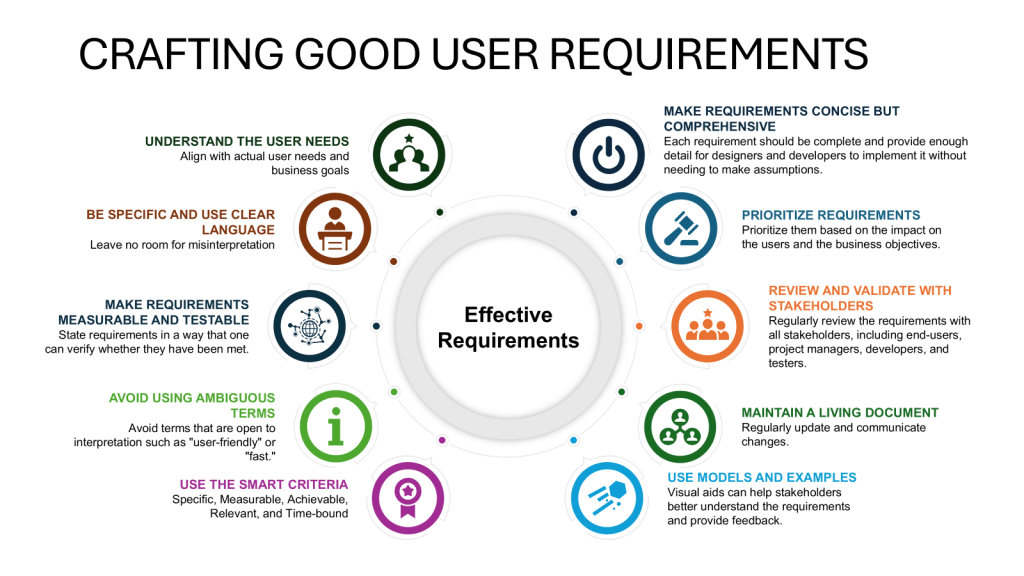
Involve multidisciplinary teams in creating the URS, including representatives from quality assurance, engineering, production, and regulatory affairs.
Conduct thorough risk assessments to identify potential GMP risks and incorporate mitigation strategies into the URS.
Ensure clear, objectively stated requirements that are verifiable during testing and commissioning.
Align the URS with company objectives and strategies to ensure long-term relevance and support.
Implement robust version control and change management processes for the URS throughout the validation lifecycle.
Executing the Control Space from the Design Space
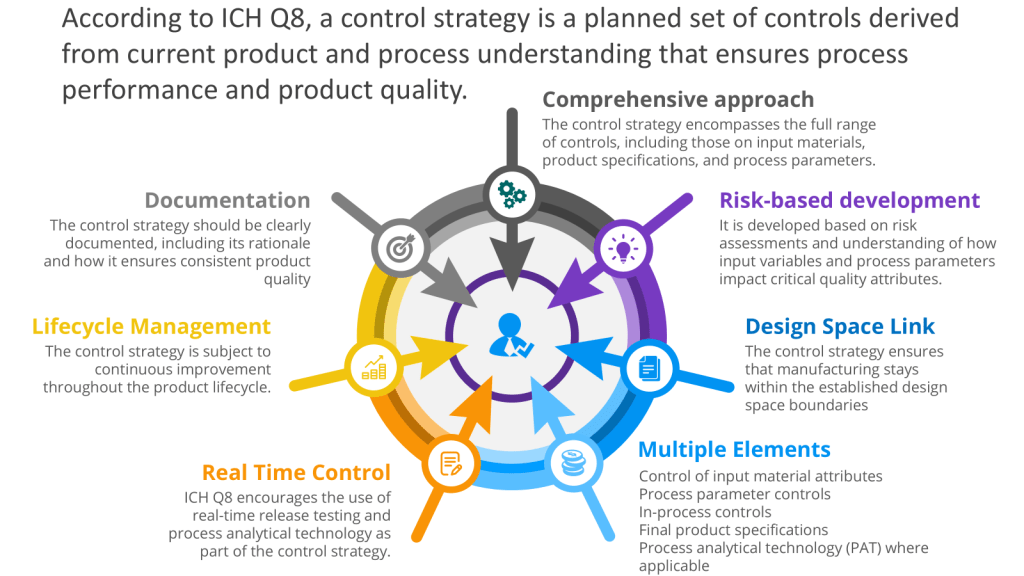
The User Requirements Specification (URS) is a mechanism for executing the control space, from the design space as outlined in ICH Q8. To understand that, let’s discuss the path from a Quality Target Product Profile (QTPP) to Critical Quality Attributes (CQAs) to Critical Process Parameters (CPPs) with Proven Acceptable Ranges (PARs), which is a crucial journey in pharmaceutical development using Quality by Design (QbD) principles. This systematic approach ensures that the final product meets the desired quality standards and user needs.
It is important to remember that this is usually a set of user requirements specifications, respecting the system boundaries.
From QTPP to CQAs
The journey begins with defining the Quality Target Product Profile (QTPP). The QTPP is a comprehensive summary of the quality characteristics that a drug product should possess to ensure its safety, efficacy, and overall quality. It serves as the foundation for product development and includes considerations such as:
Dosage strength
Delivery system
Dosage form
Container system
Purity
Stability
Sterility
Once the QTPP is established, the next step is to identify the Critical Quality Attributes (CQAs). CQAs are physical, chemical, biological, or microbiological properties that should be within appropriate limits to ensure the desired product quality. These attributes are derived from the QTPP and are critical to the safety and efficacy of the product.
From CQAs to CPPs
With the CQAs identified, the focus shifts to determining the Critical Process Parameters (CPPs). CPPs are process variables that have a direct impact on the CQAs. These parameters must be monitored and controlled to ensure that the product consistently meets the desired quality standards. Examples of CPPs include:
Temperature
pH
Cooling rate
Rotation speed
The relationship between CQAs and CPPs is established through risk assessment, experimentation, and data analysis. This step often involves Design of Experiments (DoE) to understand how changes in CPPs affect the CQAs. This is Process Characterization.
Establishing PARs
For each CPP, a Proven Acceptable Range (PAR) is determined. The PAR represents the operating range within which the CPP can vary while still ensuring that the CQAs meet the required specifications. PARs are established through rigorous testing and validation processes, often utilizing statistical tools and models.
Build the Requirements for the CPPs
The CPPs with PARs are process parameters that can affect critical quality attributes of the product and must be controlled within predetermined ranges. These are translated into user requirements. Many will specifically label these as Product User Requirements (PUR) to denote they are linked to the overall product capability. This helps to guide risk assessments and develop an overall verification approach.
Most of Us End Up on the Less than Happy Path
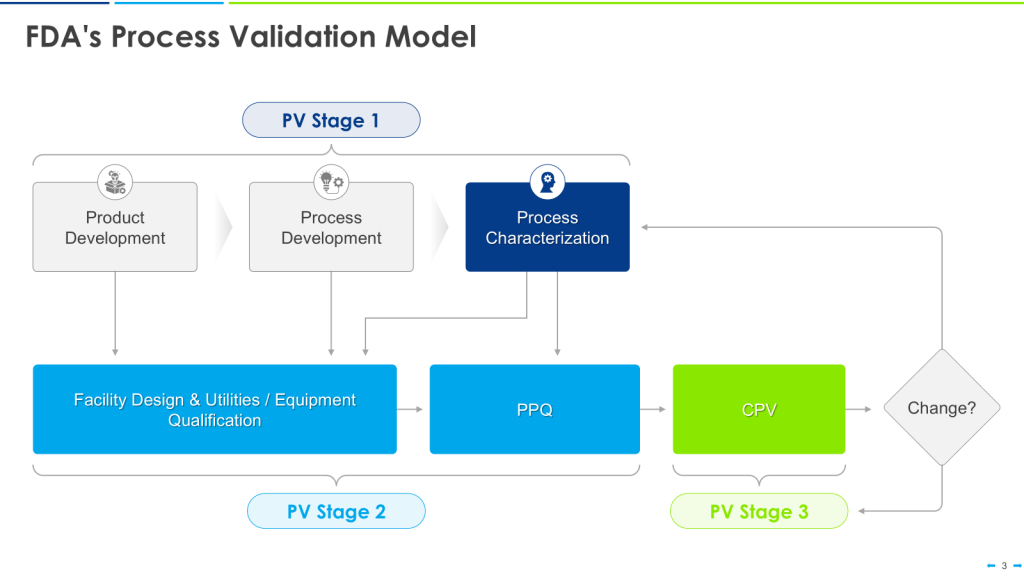
This approach is the happy path that aligns nicely with the FDA’s Process Validation Model.
This can quickly break down in the real world. Most of us go into CDMOs with already qualified equipment. We have platforms on which we’ve qualified our equipment, too. We don’t know the CPPs until just before PPQ.
This makes the user requirements even more important as living documents. Yes, we’ve qualified our equipment for these large ranges. Now that we have the CPPs, we update the user requirements for the Product User Requirements, perform an overall assessment of the gaps, and, with a risk-based approach, do additional verification activations either before or as part of Process Performance Qualification (PPQ).
I’ve been utilizing a few acronyms in a lazy way, and it is important to define them moving forward.
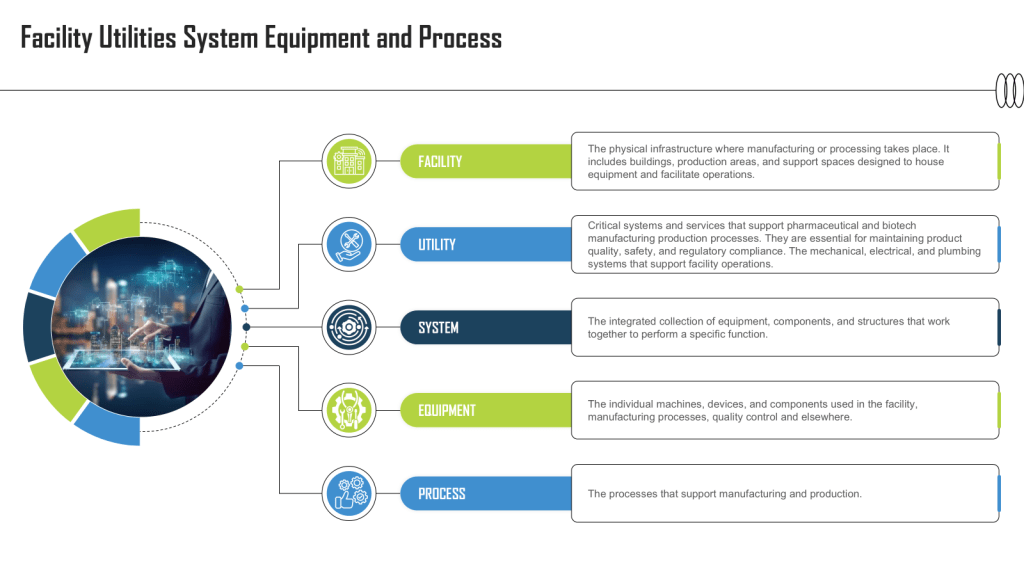
The acronyms FUSE stands for Facility Utility System Equipment; and FUSE(P) adds Process. This framework is used to describe and manage critical components of systems in facilities, particularly in industrial and pharmaceutical manufacturing settings. Here’s a breakdown of its elements:
Facility
This refers to the physical infrastructure where manufacturing or processing takes place. It includes buildings, production areas, and support spaces designed to house equipment and facilitate operations.
Utility Systems
Utilities are critical systems and services that support pharmaceutical and biotech manufacturing production processes. They are essential for maintaining product quality, safety, and regulatory compliance. The mechanical, electrical, and plumbing systems that support facility operations. Key utility systems include:
Heating, Ventilation, and Air Conditioning (HVAC)
Electrical distribution
Water systems (purified, process, and domestic)
Compressed air and gas systems
Waste management systems
System
In this context, a system refers to the integrated collection of equipment, components, and structures that work together to perform a specific function.
Equipment
This encompasses the individual machines, devices, and components used in the facility, manufacturing processes, quality control and elsewhere. Examples include mixing tanks, filling machines, packaging equipment, and quality control instruments
Process
This element refers to the manufacturing or production processes that the facility and its utility systems support. It includes:
Production workflows
Environmental control
Cleaning
Computer systems for managing manufacturing and operational processes:
The FUSE(P) framework emphasizes the interconnected nature of these elements and their collective impact on product quality, safety, and operational efficiency. It guides the design, implementation, and management of facility utility systems to ensure they meet Good Manufacturing Practice (GMP) standards and support reliable production processes.
ICH Q9(R1) emphasizes that knowledge is fundamental to effective risk management. The guideline states that “QRM is part of building knowledge and understanding risk scenarios, so that appropriate risk control can be decided upon for use during the commercial manufacturing phase.”
We need to recognize the inverse relationship between knowledge and uncertainty in risk assessment. ICH Q9(R1) notes that uncertainty may be reduced “via effective knowledge management, which enables accumulated and new information (both internal and external) to be used to support risk-based decisions throughout the product lifecycle”
In order to gauge the confidence in risk assessment we need to gauge our knowledge strength.
The Spectrum of Knowledge Strength
Knowledge strength can be categorized into three levels: weak, medium, and strong. Each level is determined by specific criteria that assess the reliability, consensus, and depth of understanding surrounding a particular subject.
Indicators of Weak Knowledge
Knowledge is considered weak if it exhibits one or more of the following characteristics:
Oversimplified Assumptions: The foundations of the knowledge rely on strong simplifications that may not accurately represent reality.
Lack of Reliable Data: There is little to no data available, or the existing information is highly unreliable or irrelevant.
Expert Disagreement: There is significant disagreement among experts in the field.
Poor Understanding of Phenomena: The underlying phenomena are poorly understood, and available models are either non-existent or known to provide inaccurate predictions.
Unexamined Knowledge: The knowledge has not been thoroughly scrutinized, potentially overlooking critical “unknown knowns.”
Hallmarks of Strong Knowledge
On the other hand, knowledge is deemed strong when it meets all of the following criteria (where relevant):
Reasonable Assumptions: The assumptions made are considered very reasonable and well-grounded.
Abundant Reliable Data: Large amounts of reliable and relevant data or information are available.
Expert Consensus: There is broad agreement among experts in the field.
Well-Understood Phenomena: The phenomena involved are well understood, and the models used provide predictions with the required accuracy.
Thoroughly Examined: The knowledge has been rigorously examined and tested.
The Middle Ground: Medium Strength Knowledge
Cases that fall between weak and strong are classified as medium strength knowledge. This category can be flexible, allowing for a broader range of scenarios to be considered strong. For example, knowledge could be classified as strong if at least one (or more) of the strong criteria are met while none of the weak criteria are present.
Strong vs Weak Knowledge
A Simplified Approach
For practical applications, a simplified version of this framework can be used:
Strong: All criteria for strong knowledge are met.
Medium: One or two criteria for strong knowledge are not met.
Weak: Three or more criteria for strong knowledge are not met.
Implications for Decision-Making
Understanding the strength of our knowledge is crucial for effective decision-making. Strong knowledge provides a solid foundation for confident choices, while weak knowledge signals the need for caution and further investigation.
When faced with weak knowledge:
Seek additional information or expert opinions
Consider multiple scenarios and potential outcomes
Implement risk mitigation strategies
When working with strong knowledge:
Make decisions with greater confidence
Focus on implementation and optimization
Monitor outcomes to validate and refine understanding
Strong knowledge typically corresponds to lower levels of uncertainty:
Level 1 Uncertainty: This aligns closely with strong knowledge, where outcomes can be estimated with reasonable accuracy within a single system model. Strong knowledge is characterized by reasonable assumptions, abundant reliable data, and well-understood phenomena, which enable accurate predictions.
Level 2 Uncertainty: While displaying alternative futures, this level still operates within a single system where probability estimates can be applied confidently. Strong knowledge often allows for this level of certainty, as it involves broad expert agreement and thoroughly examined information.
Medium Knowledge and Moderate Uncertainty (Level 3)
Medium strength knowledge often corresponds to Level 3 uncertainty:
Level 3 Uncertainty: This level involves “a multiplicity of plausible futures” with multiple interacting systems, but still within a known range of outcomes. Medium knowledge strength might involve some gaps or disagreements but still provides a foundation for identifying potential outcomes.
Weak Knowledge and Deep Uncertainty (Level 4)
Weak knowledge aligns most closely with the deepest level of uncertainty:
Level 4 Uncertainty: This level leads to an “unknown future” where we don’t understand the system and are aware of crucial unknowns. Weak knowledge, characterized by oversimplified assumptions, lack of reliable data, and poor understanding of phenomena, often results in this level of deep uncertainty.
Implications for Decision-Making
When knowledge is strong and uncertainty is low (Levels 1-2), decision-makers can rely more confidently on predictions and probability estimates.
As knowledge strength decreases and uncertainty increases (Levels 3-4), decision-makers must adopt more flexible and adaptive approaches to account for a wider range of possible futures.
The principle that “uncertainty should always be considered at the deepest proposed level” unless proven otherwise aligns with the cautious approach of assessing knowledge strength. This ensures that potential weaknesses in knowledge are not overlooked.
Conclusion
By systematically evaluating the strength of our knowledge using this framework, we can make more informed decisions, identify areas that require further investigation, and better understand the limitations of our current understanding. Remember, the goal is not always to achieve perfect knowledge but to recognize the level of certainty we have and act accordingly.