Predicts compliance with FDA 21 CFR 211.100 (process control)
FDA 21 CFR 211, ICH Q10, ICH Q9
Lagging
Average Time to Close Change Requests
Validates efficiency of change implementation (EudraLex Annex 15)
EU GMP Annex 15
KRI
Leading
Unresolved CAPAs Linked to Change Requests
Identifies systemic risks before deviations occur (FDA Warning Letters)
21 CFR 211.22, ICH Q7
Lagging
Repeat Deviations Post-Change
Reflects failure to address root causes (FDA 483 Observations)
21 CFR 211.192
KBI
Leading
Cross-Functional Review Participation Rate
Encourages proactive collaboration in change evaluation
ICH Q10 Section 3.2.3
Lagging
Reduction in Documentation Errors Post-Training
Validates effectiveness of staff competency programs
EU 1252/2014 Article 14
Key Performance Indicators (KPIs)
Leading KPI:
Change Requests with Completed Risk Assessments: Measures proactive compliance with FDA requirements for risk-based change evaluation. A rate <90% triggers quality reviews.
Lagging KPI:
Time to Close Changes: Benchmarks against EMA’s 30-day resolution expectation for critical changes. Prolonged closure (>45 days) indicates process bottlenecks.
Key Risk Indicators (KRIs)
Leading KRI:
Unresolved CAPAs: Predicts validation gaps; >5 open CAPAs per change violates FDA’s “state of control” mandate.
Lagging KRI:
Repeat Deviations: >3 repeat deviations quarterly triggers mandatory revalidation per FDA 21 CFR 211.180.
Documentation Errors: Post-training error reduction <30% prompts requalification under EU GMP Chapter 4.
Implementation Guidance
Align with Regulatory Thresholds: Set leading KPI targets using FDA’s 2025 draft guidance: ≥95% risk assessment completion for high-impact changes.
Automate Tracking: Integrate metrics with eQMS software to monitor CAPA aging (leading KRI) and deviation trends (lagging KRI) in real time.
Link to Training: Tie lagging KBIs to annual GMP refresher courses, as required by EU 1252/2014 Article 14.
Why It Matters: Leading metrics enable proactive mitigation of change-related risks (e.g., unresolved CAPAs predicting audit failures), while lagging metrics validate adherence to FDA’s lifecycle approach for process validation. Balancing both ensures compliance with 21 CFR 211’s “state of control” mandate while fostering continuous improvement.
The folks behind GAMP5 are perhaps the worst in naming things. And one of the worse is the whole standard versus normal changes. Maybe when naming two types of changes do not use strong synonyms. Seems like good advice in general, when naming categories don’t draw from a list of synonyms.
So a standard change is one that is always done the same way, can be proceduralized, and is of low risk. In exchange for doing all that work, you get to do them by a standard process without the evaluation of a GxP change control, because you have already done all the evaluation and the implementation is the same every single time. If you need to perform evaluation or create an action plan, it is not a standard change.
Normal Change
Any change that is not a Standard change or Emergency change.
Requires full Change Management review for each occurrence.
Raised as a GxP Change Control.
Approved or rejected by the Change Manager, which usually means Quality review.
Often involves non-trivial changes to services, processes, or infrastructure.
May require somewhat unique or novel approaches.
Undergoes assessment and action planning.
The key distinction is that Standard changes have pre-approved processes and do not require individual approval, while Normal changes go through the full change management process each time. Standard changes are meant for routine, low-risk activities, while Normal changes are for more significant modifications that require careful review and approval.
What About Emergency Changes
An emergency change is a change that must be implemented immediately to address an unexpected situation that requires urgent action to:
Ensure continued operations
Address a critical issue or crisis
Key characteristics of emergency changes in GAMP 5:
They need to be expedited quickly to obtain authorization and approval before implementation.
They follow a fast-track process compared to normal changes.
A full change control should be filed for evaluation within a few business days after execution.
Impacted items are typically withheld from further use pending evaluation of the emergency change.
They represent a situation where there is an acceptable level of risk expected due to the urgent nature.
Specific approvals and authorizations are still required, but through an accelerated process.
Emergency changes may not be as thoroughly tested as normal changes due to time constraints.
A remediation or back-out process should be included in case issues arise from the rapid implementation.
The goal is to address the critical situation while minimizing impact to live services.
The key difference from standard or normal changes is that emergency changes follow an expedited process to deal with urgent, unforeseen issues that require immediate action, while still maintaining some level of control and documentation. However, they should still be evaluated and fully documented after implementation.
It is crucial for a Marketing Authorization Holder (MAH) to review and approve changes made by a Contract Development and Manufacturing Organization (CDMO) for several important reasons:
Regulatory Compliance
The Market Authorization Holder (MAH) – or the sponsor for pre-commercial GMP manufacturing – bears the primary responsibility for ensuring compliance with the marketing authorization and regulatory requirements throughout the product’s lifecycle. By reviewing and approving CDMO changes, the MAH can:
Ensure changes align with the approved marketing authorization
Verify that any variations to the marketing authorization are properly submitted to regulatory authorities
Maintain oversight of post-approval change management as required by regulations
Before I go any further on the topic I want you to go and read my post Classification of Changes for GMP/GDP. This post will build on that discussion.
I think it is better for the CDMO to put a lot of thought into this, and the MAH (the client) to evaluate and adapt. For all but the big players, the volume is going to be on the CDMO’s side. But if you are the client and your CDMO hasn’t taken this into account to the appropriate degree, you need to ensure appropriate steps taken. As such the rest of this post will be written from the CDMO’s side, but the same principles apply to the MAH (and should be included in the audit program).
Remember we have three goals:
Fulfill our contractual responsibilities
Help the MAH maintain appropriate control as the product owner
Ensure alignment between both parties on change implementation
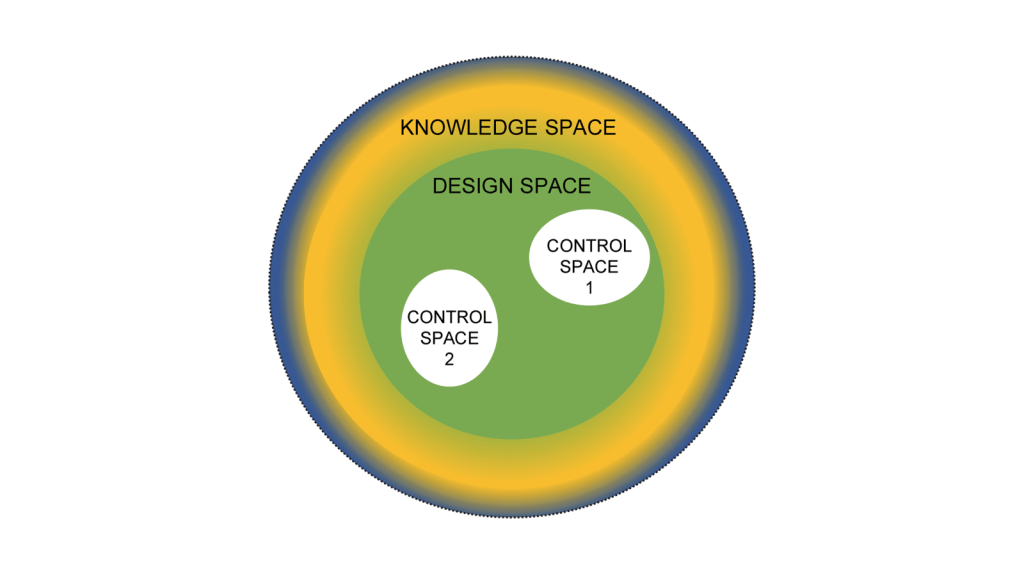
The critical requirement here is ensuring the right changes get to the right client so they can be filled the right way. Returning to basics, we are approaching changes as:
Now it’s easy to apply this to product. Create and/or receive the design space and the control space. Everything that falls into a non-established condition does not get reported to the client at time of execution. If it is “Do and Report” is is in the APQR. If it is “Do and Record” they can see it during the audit.
Where a lot of CDMOs trip up here are facility and quality system changes. My recommendation here is the same, define a design space based on the CMC section of the Common Technical Document which basically boils down to:
The CMC (Chemistry, Manufacturing, and Controls) section of a regulatory dossier typically includes the following key facility-related information:
Manufacturing Facilities
Names and addresses of all manufacturing, testing, and storage facilities involved in production
Description of the manufacturing operations performed at each site
Floor plans and layouts of production areas
Details on utilities and support systems (HVAC, water, gases, etc.)
Information on facility design features for contamination control and product protection
Equipment
List of major production and laboratory equipment
Equipment specifications and capacities
Cleaning and maintenance procedures for equipment
Environmental Controls
Description of clean room classifications and environmental monitoring programs
Air handling systems and controls
Water systems (purified water, water for injection) and controls
Material Flow
Personnel and material flow diagrams
Segregation of operations to prevent cross-contamination
Quality Control Laboratories
Description of QC lab facilities and equipment
Environmental controls in QC labs
Storage Areas
Description of storage facilities for raw materials, intermediates, and finished products
Storage conditions and controls (temperature, humidity, etc.)
There is a whole lot of wiggle room here in things that fall into “Do and Record.” By building this into your change control system you can delineate what goes to to the client and what doesn’t. I recommend sitting down with this list and deciding what types of changes fall into “Tell and Do” – what you ask permission from clients before doing; “Do and Report” – what goes in the APQR; and, “Do and Record” – what the client sees when they audit.
You know have good rules on what changes go to a client for prior approval and which ones do not. This gets codified in two places: the change control process and the quality/technical agreement.
Some other things to build into your change control process:
Documenting when a client requests a change, the reason and the impact on the platform. Remember you have other clients, and more and more CDMO’s are offering a platform, so there needs to be appropriate review and endorsement.
Think through how changes to facility (and other platform elements) are communicated and gated for multiple clients. Have a mechanism to manage client specific activities and to track first-product impacted for multiple products.
Have clear timelines and expectations on change communication and approval with the client in the quality/technical agreement. Hold each other accountable.
Have contingency plans. There will always be that one client who will be in shortage if you make that urgent change just when you want/need to.
Have a method for evaluating requested changes to the change plan by clients and making decisions around it. There will be that one client who doesn’t agree or wants something weird that disagrees with what all the other clients want.
Have rules in place to manage changes inactive for long periods or extensions specific for those changes that rise to client approval. These will have a different flow than internal changes.
I’ve used a bit of commercial headspace for this post, relying on the APQR. For clinical processes, product tends to fall into campaign-mindset, so “Do and Report” ends up being more a clinical campaign change report than an APQR.
Classification of change controls within change management is a common and widely accepted best practice. It stems from the requirement that change proposals as assessed from a risk perspective, where:
the level of rigor, effort and documentation is commensurate with the level of risk,
the risk assessments adequately evaluate the potential risks and benefits of changes to product quality, safety and efficacy, and
those risk assessments consider the potential risks and benefits to other products, processes and systems.
Classification for GMP/GDP changes itself is not a requirement, it is a guidance, best found in the PIC/S Recommendation “How to Evaluate and Demonstrate the Effectiveness of a Pharmaceutical Quality System in relation to Risk-based Change Management” (PI 054-1) which states in section 5.2 “Change Management procedures often require a risk-based classification (e.g. critical, major, minor) to be assigned to proposed changes as well as an impact assessment to be performed. The latter routinely determines the potential impacts of the proposed change on various items, such as product quality, documentation, cleaning, maintenance, regulatory compliance, etc. In some cases, especially for simple and minor/low risk changes, an impact assessment is sufficient to document the risk-based rationale for a change without the use of more formal risk assessment tools or approaches.”
The PIC/S tells us that these categories drive the amount of rigor a change control requires, which is a great reason to have them. We spend time creating and confirming our categories, and then we only need to perform more rigorous risk assessments on the big changes.
How should we build this risk-based classification system? There are four criteria that drive this:
Potential regulatory impact
Potential impact on the qualified and validated state
Potential impact on the ability to disposition and ship product
Complexity
I tend to use only two categories, defined like this:
Major has Significant Impact: Changes that have a considerable potential impact on the process, product quality, safety, or regulatory status.
Minor has Limited Impact: Changes that have minimal or no significant impact on the process, product quality, safety, or regulatory status.
For regulatory impact, it really is as easy as dividing things into the four categories. “Do, Report, and Do and Record are minors. “Do and Tell” are majors, and “Tell and Do are either majors or critical based on how you slice it.
When considering potential validation impact you’ll leverage your process risk assessments and your validated state to determine what is in that bucket. This is why I like a document like an operational control strategy because this tells me exactly what impacts my validated state and I can just it to form this category.
The potential impact on the ability to disposition and ship the product has me looking at what can impact the ability to release and get the product out the door, which is an important aspect of what we do. Remember, a shortage of products is a quality issue.
Complexity looks at how many processes and systems are impacted and how many functions and areas are involved. The more complex, the more formal risk assessment is required. For example, you might use groupings like this:
Low level of complexity
Requires actions from the change owner and the system owner’s department(s) only
Impacts 1 system
<10 document revisions (approximate)
<2 potential training audiences (approximate)
Higher complexity:
Requires actions from more than change owner and system owner
Impacts more than one system
>10 document revisions
>2 potential training audiences (approximate)
The where of making the classification also makes a difference. I recommend up front, agreed to by the change owner and quality and it then drives everything. Doing it just before approval really just decides who gets to approve the change control and whether it goes to CCRB or not.
These classifications can be loose guidelines; for example, a table that looks at the first three categories and then by complexity. Your rating depends on whichever Impact or Complexity is higher.
Impact of Change (regulatory, validation, product)
Complexity of Change
Minor
No risk to patient as assessed by SISPQ, product, or validated equipment or process AND No regulatory impact.
Limited impact to only one system/functional area AND Has defined process for implementation of change. (e.g. all action items are per defined procedures)
Major
Potential impact to patient or product SISPQ or validated equipment or process or compliance
Impacts multiple systems / functional areas OR Has defined process for implementation of change
Critical
High likelihood of impact to patient, product SISPQ or validated equipment or process or compliance
Impacts multiple systems / functional areas OR Implementation activities are not pre-defined or governed by formal internal system
Or we could try for something much more specific. The advantage of specific is any change owner can start making the determination. Something like this:
Change Category
Change Description
Manufacturing Processes
In-process labeling
Changes to Process Control and Operating Parameters (tightening/shifting) within current batch record (does not impact established conditions)
The addition of in-process or final product samples
Changes to sample volume for in-process or finished product samples
Addition of new ancillary equipment (e.g. no product contact, does not control process steps) to the process
Analytical Methods
Changes to the qualification of a critical reagent (i.e., in-house produced assay standards and controls)
Use of an additional new instrument of the identical model and vendor
Change in compendial method to comply with formal updates to compendia, provided it does not involve the widening of system suitability or acceptance criteria
Equipment/instruments calibration, maintenance, and cleaning
Changes to software or validated analytical spreadsheets that do not impact the current validated state of the method
Movement of instruments from one location to another in the same room/lab
Initial validation of analytical spreadsheets for use in calculation of data and results defined by a specific analytical method, provided it does not replace a worksheet in an SOP (if so, this change may be reportable)
Changes to non-critical equipment or materials that allow “or equivalent” in current method, provided method re-validation is not required
Drug Substance or Drug Product Specifications/ Limits
Changes to the sampling plan involving changes to the number of extra samples or amount of sample provided to QC or CMO as appropriate.
Changes to the storage and/or shipping conditions of samples (except for stability vials)
Raw Materials/Com ponents
Compendial Specification Changes to meet Compendial updates
Non-product contact filters
Vendor increase or decrease in the number of items per shipping container, or the size of the shipping or outer container
Changes to the vendor Certificate of Analysis (format change only)
Changes in recommended expiration date and/or storage conditions of raw material
Finished Goods
Catalog Number changes to components
Creation of label at contract manufacturing site for existing presentation (assuming ‘No’ other change to already approved label)
Changing position of pharmacode on leaflet
Computer
When there is no validation impact
Facility, Utilities, Systems and Equipment (including Automation)
Equipment/instrument maintenance
Decommissioning of equipment not classified as critical equipment
Computer programming that affects non-production equipment
Alarms (i.e., notification system for out of tolerances)
Cleaning and Sanitization of Manufacturing facilities and non-product Contact equipment
Upgrade of Application Software or operating system
Alarm set point changes
Creating user groups and modifying user group privileges
Tuning parameter, adjustment to the gain, reset and rate of a PID controller
Phase or sequence change that does not affect the function and performance
Modifying a phase prompt or message (technical change)
Addition of a graphic, adding or changing a non-static device to a graphic (technical change)
Addition or changing to an interlock/permissive trigger
Changes to alarm paging/notification functionality
Spend the time on your classification structure. You will use it to:
Determine level of risk assessment (major yes, minor no)
Determine approvals (minors can be as simple as change owner and quality)
Does this change require a CCRB? Only send majors.
Potential risks with the current state (until changes are implemented) and any risks that might be temporarily introduced during the change process are adequately assessed.
Interim controls (short-term measures), as needed, are identified and implemented in a timely manner to monitor/mitigate risks associated with the current situation (until change implementation).
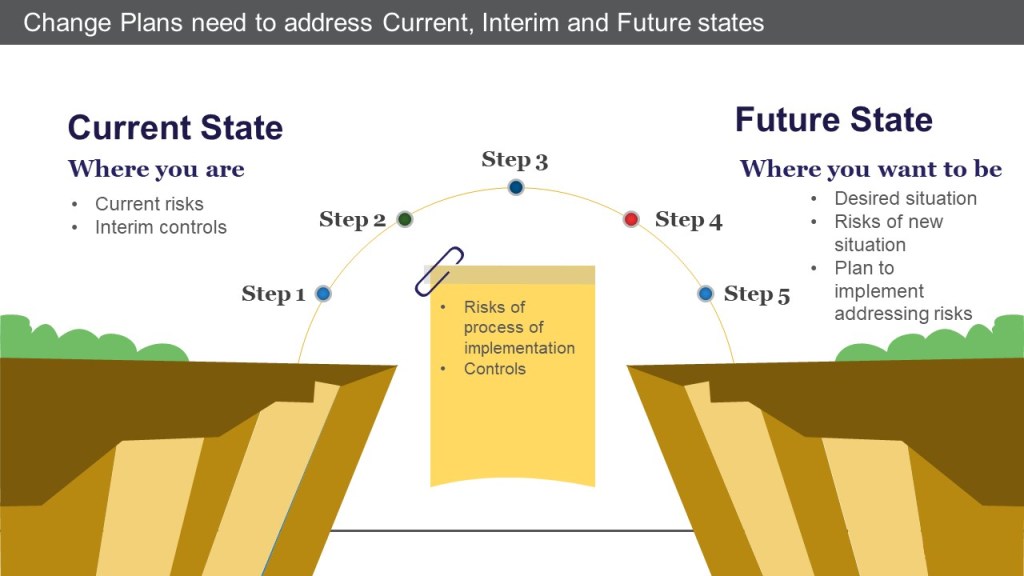
This section also recognizes that changes introduce an interim set of conditions that lead to implementation – from opening a machine through massive construction activities, and just about anything else.
A good change control does not just address what is necessary to implement the desired future state. It also contains interim controls for managing the current state until the future is implemented, and addresses all of the potential risks during the process of implementation.
Change plans address current, interim, and future states
Often the change control stems from an action plan in a CAPA record, where the interim controls are specifically called out and detailed. These may be adequate for a relatively brief period but are not realistically sustainable. Remember that these interim controls must also go through the appropriate change control process. Which may have been a separate change control or be part of this change control. In all cases, the change control should either determine that the CAPA plan is adequate or identify additional risks that were identified during change planning.
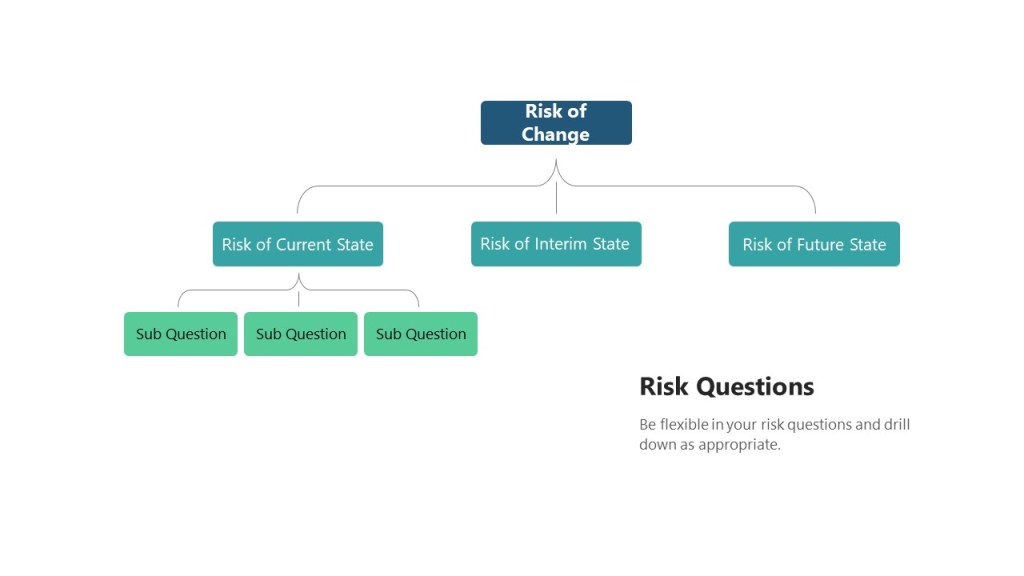
Risk assessments for change control should really have three basic risk questions:
What are the risks of the current state as we implement the change?
What are the risks of the implementation process?
What are the risks of the future state?
As discussed elsewhere, these questions are really branching trees.
The Risk Question branches in changes
Interim controls in a change are usually little temporary changes in the change plan that either mitigate occurrence or impact.
Interim Control Type
Mitigate Occurrence
Mitigate Impact
Examples
Every record processed will be reviewed for the event error before completion. Revised preventive maintenance procedure to require vibration test at next preventive maintenance. Increase cleaning frequency of incubator.
Add emergency response steps to address a valve that is malfunctioning. Add engineering check for pump prior to each use. Perform cleaning log review prior to use.
As temporary changes, it is important to determine how they will be implemented, if they require monitoring, and how they will go away. Often the process of implementation of the change removes the temporary changes, but that is not always the case.