The environment for commissioning, qualification, and validation (CQV) professionals remains defined by persistent challenges. Rapid technological advancements—most notably in artificial intelligence, machine learning, and automation—are constantly reshaping the expectations for validation. Compliance requirements are in frequent flux as agencies modernize guidance, while the complexity of novel biologics and therapies demands ever-higher standards of sterility, traceability, and process control. The shift towards digital systems has introduced significant hurdles in data management and integration, often stretching already limited resources. At the same time, organizations are expected to fully embrace risk-based, science-first approaches, which require new methodologies and skills. Finally, true validation now hinges on effective collaboration and knowledge-sharing among increasingly cross-functional and global teams.
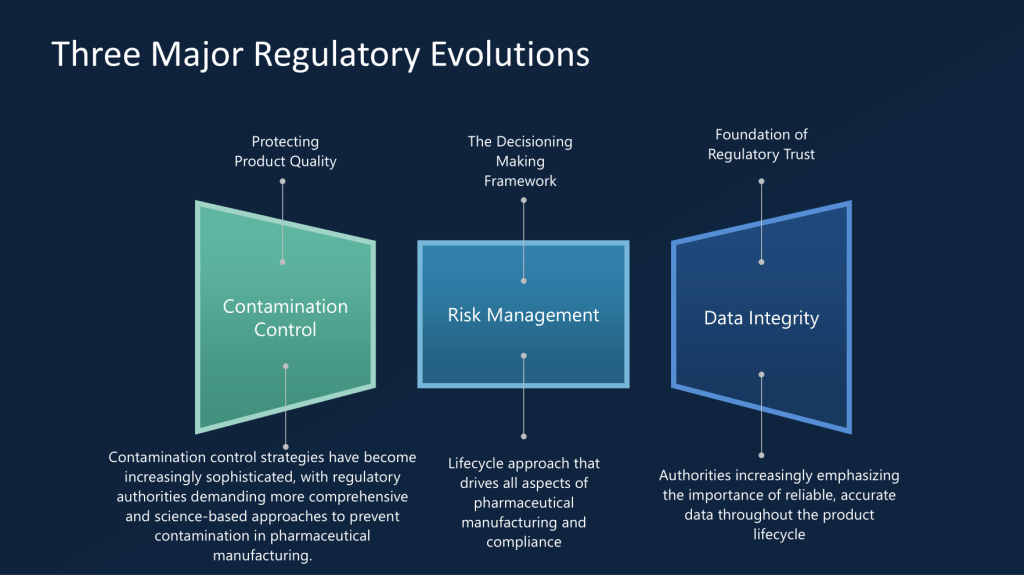
Overlaying these challenges, three major regulatory paradigm shifts are transforming the expectations around risk management, contamination control, and data integrity. Data integrity in particular has become an international touchpoint. Since the landmark PIC/S guidance in 2021 and matching World Health Organization updates, agencies have made it clear that trustworthy, accurate, and defendable data—whether paper-based or digital—are the foundation of regulatory confidence. Comprehensive data governance, end-to-end traceability, and robust documentation are now all non-negotiable.
Contamination control is experiencing its own revolution. The August 2023 overhaul of EU GMP Annex 1 set a new benchmark for sterile manufacturing. The core concept, the Contamination Control Strategy (CCS), formalizes expectations: every manufacturer must systematically identify, map, and control contamination risks across the entire product lifecycle. From supply chain vigilance to environmental monitoring, regulators are pushing for a proactive, science-driven, and holistic approach, far beyond previous practices that too often relied on reactive measures. We this reflected in recent USP drafts as well.
Quality risk management (QRM) also has a new regulatory backbone. The ICH Q9(R1) revision, finalized in 2023, addresses long-standing shortcomings—particularly subjectivity and lack of consistency—in how risks are identified and managed. The European Medicines Agency’s ongoing revision of EudraLex Chapter 1, now aiming for finalization in 2026, will further require organizations to embed preventative, science-based risk management within globalized and complex supply chain operations. Modern products and supply webs simply cannot be managed with last-generation compliance thinking.
The EU Digital Modernization: Chapter 4, Annex 11, and Annex 22
With the rapid digitalization of pharma, the European Union has embarked on an ambitious modernization of its GMP framework. At the heart of these changes are the upcoming revisions to Chapter 4 (Documentation), Annex 11 (Computerised Systems), and the anticipated implementation of Annex 22 (Artificial Intelligence).
Chapter 4—Documentation is being thoroughly updated in parallel with Annex 11. The current chapter, which governs all aspects of documentation in GMP environments, was last revised in 2011. Its modernization is a direct response to the prevalence of digital tools—electronic records, digital signatures, and interconnected documentation systems. The revised Chapter 4 is expected to provide much clearer requirements for the management, review, retention, and security of both paper and electronic records, ensuring that information flows align seamlessly with the increasingly digital processes described in Annex 11. Together, these updates will enable companies to phase out paper where possible, provided electronic systems are validated, auditable, and secure.
Annex 11—Computerised Systems will see its most significant overhaul since the dawn of digital pharma. The new guidance, scheduled for publication and adoption in 2026, directly addresses areas that the previous version left insufficiently covered. The scope now embraces the tectonic shift toward AI, machine learning, cloud-based services, agile project management, and advanced digital workflows. For instance, close attention is being paid to the robustness of electronic signatures, demanding multi-factor authentication, time-zoned audit trails, and explicit provisions for non-repudiation. Hybrid (wet-ink/digital) records will only be acceptable if they can demonstrate tamper-evidence via hashes or equivalent mechanisms. Especially significant is the regulation of “open systems” such as SaaS and cloud platforms. Here, organizations can no longer rely on traditional username/password models; instead, compliance with standards like eIDAS for trusted digital providers is expected, with more of the technical compliance burden shifting onto certified digital partners.
The new Annex 11 also calls for enhanced technical controls throughout computerized systems, proportional risk management protocols for new technologies, and a far greater emphasis on continuous supplier oversight and lifecycle validation. Integration with the revised Chapter 4 ensures that documentation requirements and data management are harmonized across the digital value chain.
The introduction of Annex 22 represents a pivotal moment in the regulatory landscape for pharmaceutical manufacturing in Europe. This annex is the EU’s first dedicated framework addressing the use of Artificial Intelligence (AI) and machine learning in the production of active substances and medicinal products, responding to the rapid digital transformation now reshaping the industry.
Annex 22 sets out explicit requirements to ensure that any AI-based systems integrated into GMP-regulated environments are rigorously controlled and demonstrably trustworthy. It starts by mandating that manufacturers clearly define the intended use of any AI model deployed, ensuring its purpose is scientifically justified and risk-appropriate.
Quality risk management forms the backbone of Annex 22. Manufacturers must establish performance metrics tailored to the specific application and product risk profile of AI, and they are required to demonstrate the suitability and adequacy of all data used for model training, validation, and testing. Strong data governance principles apply: manufacturers need robust controls over data quality, traceability, and security throughout the AI system’s lifecycle.
The annex foresees a continuous oversight regime. This includes change control processes for AI models, ongoing monitoring of performance to detect drift or failures, and formally documented procedures for human intervention where necessary. The emphasis is on ensuring that, even as AI augments or automates manufacturing processes, human review and responsibility remain central for all quality- and safety-critical steps.
By introducing these requirements, Annex 22 aims to provide sufficient flexibility to enable innovation, while anchoring AI applications within a robust regulatory framework that safeguards product quality and patient safety at every stage. Together with the updates to Chapter 4 and Annex 11, Annex 22 gives companies clear, actionable expectations for responsibly harnessing digital innovation in the manufacturing environment.
Life Cycle Integration, Analytical Validation, and AI/ML Guidance
Across global regulators, a clear consensus has taken shape: validation must be seen as a continuous lifecycle process, not as a “check-the-box” activity. The latest WHO technical reports, the USP’s evolving chapters (notably <1058> and <1220>), and the harmonized ICH Q14 all signal a new age of ongoing qualification, continuous assurance, change management, and systematic performance verification. The scope of validation stretches from the design qualification stage through annual review and revalidation after every significant change.
A parallel wave of guidance for AI and machine learning is cresting. The EMA, FDA, MHRA, and WHO are now releasing coordinated documents addressing everything from transparent model architecture and dataset controls to rigorous “human-in-the-loop” safeguards for critical manufacturing decisions, including the new draft Annex 22. Data governance—traceability, security, and data quality—has never been under more scrutiny.
Regulatory Body
Document Title
Publication Date
Status
Key Focus Areas
EMA
Reflection Paper on the Use of Artificial Intelligence in the Medicinal Product Lifecycle
Oct-24
Final
Risk-based approach for AI/ML development, deployment, and performance monitoring across product lifecycle including manufacturing
EMA/HMA
Multi-annual AI Workplan 2023-2028
Dec-23
Final
Strategic framework for European medicines regulatory network to utilize AI while managing risks
EMA
Annex 22 Artificial Intelligence
Jul-25
Draft
Establishes requirements for the use of AI and machine learning in the manufacturing of active substances and medicinal products.
FDA
Considerations for the Use of AI to Support Regulatory Decision Making for Drug and Biological Products
Feb-25
Draft
Guidelines for using AI to generate information for regulatory submissions
FDA
Discussion Paper on AI in the Manufacture of Medicines
May-23
Published
Considerations for cloud applications, IoT data management, regulatory oversight of AI in manufacturing
FDA/Health Canada/MHRA
Good Machine Learning Practice for Medical Device Development Guiding Principles
Mar-25
Final
10 principles to inform development of Good Machine Learning Practice
WHO
Guidelines for AI Regulation in Health Care
Oct-23
Final
Six regulatory areas including transparency, risk management, data quality
MHRA
AI Regulatory Strategy
Apr-24
Final
Strategic approach based on safety, transparency, fairness, accountability, and contestability principles
EFPIA
Position Paper on Application of AI in a GMP Manufacturing Environment
Sep-24
Published
Industry position on using existing GMP framework to embrace AI/ML solutions
The Time is Now
The world of validation is no longer controlled by periodic updates or leisurely transitions. Change is the new baseline. Regulatory authorities have codified the digital, risk-based, and globally harmonized future—are your systems, people, and partners ready?
In the highly regulated pharmaceutical industry, ensuring the quality, safety, and efficacy of products is paramount. Two critical components of pharmaceutical quality management are Quality Assurance (QA) and Quality Control (QC). While these terms are sometimes used interchangeably, they represent distinct approaches with different focuses, methodologies, and objectives within pharmaceutical manufacturing. Understanding the differences between QA and QC is essential for pharmaceutical companies to effectively manage their quality processes and meet regulatory requirements.
Quality Assurance (QA) and Quality Control (QC) are both essential and complementary pillars of pharmaceutical quality management, each playing a distinct yet interconnected role in ensuring product safety, efficacy, and regulatory compliance. QA establishes the systems, procedures, and preventive measures that form the foundation for consistent quality throughout the manufacturing process, while QC verifies the effectiveness of these systems by testing and inspecting products to ensure they meet established standards. The synergy between QA and QC creates a robust feedback loop: QC identifies deviations or defects through analytical testing, and QA uses this information to drive process improvements, update protocols, and implement corrective and preventive actions. This collaboration not only helps prevent the release of substandard products but also fosters a culture of continuous improvement, risk mitigation, and regulatory compliance, making both QA and QC indispensable for maintaining the highest standards in pharmaceutical manufacturing.
Definition and Scope
Quality Assurance (QA) is a comprehensive, proactive approach focused on preventing defects by establishing robust systems and processes throughout the entire product lifecycle. It encompasses the totality of arrangements made to ensure pharmaceutical products meet the quality required for their intended use. QA is process-oriented and aims to build quality into every stage of development and manufacturing.
Quality Control (QC) is a reactive, product-oriented approach that involves testing, inspection, and verification of finished products to detect and address defects or deviations from established standards. QC serves as a checkpoint to identify any issues that may have slipped through the manufacturing process.
Approach: Proactive vs. Reactive
One of the most fundamental differences between QA and QC lies in their approach to quality management:
QA takes a proactive approach by focusing on preventing defects and deviations before they occur. It establishes robust quality management systems, procedures, and processes to minimize the risk of quality issues.
QC takes a reactive approach by focusing on detecting and addressing deviations and defects after they have occurred. It involves testing, sampling, and inspection activities to identify non-conformities and ensure products meet established quality standards.
Focus: Process vs. Product
QA is process-oriented, focusing on establishing and maintaining robust processes and procedures to ensure consistent product quality. It involves developing standard operating procedures (SOPs), documentation, and validation protocols.
QC is product-oriented, focusing on verifying the quality of finished products through testing and inspection. It ensures that the final product meets predetermined specifications before release to the market.
Comparison Table: QA vs. QC in Pharmaceutical Manufacturing
Aspect
Quality Assurance (QA)
Quality Control (QC)
Definition
A comprehensive, proactive approach focused on preventing defects by establishing robust systems and processes
A reactive, product-oriented approach that involves testing and verification of finished products
Focus
Process-oriented, focusing on how products are made
Product-oriented, focusing on what is produced
Approach
Proactive – prevents defects before they occur
Reactive – detects defects after they occur
Timing
Before and during production
During and after production
Responsibility
Establishing systems, procedures, and documentation
To build quality into every stage of development and manufacturing
To identify non-conformities and ensure products meet specifications
Methodology
Establishing SOPs, validation protocols, and quality management systems
Sampling, testing, inspection, and verification activities
Scope
Spans the entire product lifecycle from development to discontinuation
Primarily focused on manufacturing and finished products
Relationship to GMP
Ensures GMP implementation through systems and processes
Verifies GMP compliance through testing and inspection
The Quality Continuum: QA and QC as Complementary Approaches
Rather than viewing QA and QC as separate entities, modern pharmaceutical quality systems recognize them as part of a continuous spectrum of quality management activities. This continuum spans the entire product lifecycle, from development through manufacturing to post-market surveillance.
The Integrated Quality Approach
QA and QC represent different points on the quality continuum but work together to ensure comprehensive quality management. The overlap between QA and QC creates an integrated quality approach where both preventive and detective measures work in harmony. This integration is essential for maintaining what regulators call a “state of control” – a condition in which the set of controls consistently provides assurance of continued process performance and product quality.
Quality Risk Management as a Bridge
Quality Risk Management (QRM) serves as a bridge between QA and QC activities, providing a systematic approach to quality decision-making. By identifying, assessing, and controlling risks throughout the product lifecycle, QRM helps determine where QA preventive measures and QC detective measures should be applied most effectively.
The concept of a “criticality continuum” further illustrates how QA and QC work together. Rather than categorizing quality attributes and process parameters as simply critical or non-critical, this approach recognizes varying degrees of criticality that require different levels of control and monitoring.
Organizational Models for QA and QC in Pharmaceutical Companies
Pharmaceutical companies employ various organizational structures to manage their quality functions. The choice of structure depends on factors such as company size, product portfolio complexity, regulatory requirements, and corporate culture.
Common Organizational Models
Integrated Quality Unit
In this model, QA and QC functions are combined under a single Quality Unit with shared leadership and resources. This approach promotes streamlined communication and a unified approach to quality management. However, it may present challenges related to potential conflicts of interest and lack of independent verification.
Separate QA and QC Departments
Many pharmaceutical companies maintain separate QA and QC departments, each with distinct leadership reporting to a higher-level quality executive. This structure provides clear separation of responsibilities and specialized focus but may create communication barriers and resource inefficiencies.
QA as a Standalone Department, QC Integrated with Operations
In this organizational model, the Quality Assurance (QA) function operates as an independent department, while Quality Control (QC) is grouped within the same department as other operations functions, such as manufacturing and production. This structure is designed to balance independent oversight with operational efficiency.
Centralized Quality Organization
Large pharmaceutical companies often adopt a centralized quality organization where quality functions are consolidated at the corporate level with standardized processes across all manufacturing sites. This model ensures consistent quality standards and efficient knowledge sharing but may be less adaptable to site-specific needs.
Decentralized Quality Organization
In contrast, some companies distribute quality functions across manufacturing sites with site-specific quality teams. This approach allows for site-specific quality focus and faster decision-making but may lead to inconsistent quality practices and regulatory compliance challenges.
Matrix Quality Organization
A matrix quality organization combines elements of both centralized and decentralized models. Quality personnel report to both functional quality leaders and operational/site leaders, providing a balance between standardization and site-specific needs. However, this structure can create complex reporting relationships and potential conflicts in priorities.
The Quality Unit: Overarching Responsibility for Pharmaceutical Quality
Concept and Definition of the Quality Unit
The Quality Unit is a fundamental concept in pharmaceutical manufacturing, representing the organizational entity responsible for overseeing all quality-related activities. According to FDA guidance, the Quality Unit is “any person or organizational element designated by the firm to be responsible for the duties relating to quality control”.
The concept of a Quality Unit was solidified in FDA’s 2006 guidance, “Quality Systems Approach to Pharmaceutical Current Good Manufacturing Practice Regulations,” which defined it as the entity responsible for creating, monitoring, and implementing a quality system.
Independence and Authority of the Quality Unit
Regulatory agencies emphasize that the Quality Unit must maintain independence from production operations to ensure objective quality oversight. This independence is critical for the Quality Unit to fulfill its responsibility of approving or rejecting materials, processes, and products without undue influence from production pressures.
The Quality Unit must have sufficient authority and resources to carry out its responsibilities effectively. This includes the authority to investigate quality issues, implement corrective actions, and make final decisions regarding product release.
How QA and QC Contribute to Environmental Monitoring and Contamination Control
Environmental monitoring (EM) and contamination control are critical pillars of pharmaceutical manufacturing quality systems, requiring the coordinated efforts of both Quality Assurance (QA) and Quality Control (QC) functions. While QA focuses on establishing preventive systems and procedures, QC provides the verification and testing that ensures these systems are effective. Together, they create a comprehensive framework for maintaining aseptic manufacturing environments and protecting product integrity. This also serves as a great example of the continuum in action.
QA Contributions to Environmental Monitoring and Contamination Control
System Design and Program Development
Quality Assurance takes the lead in establishing the foundational framework for environmental monitoring programs. QA is responsible for designing comprehensive EM programs that include sampling plans, alert and action limits, and risk-based monitoring locations. This involves developing a systematic approach that addresses all critical elements including types of monitoring methods, culture media and incubation conditions, frequency of environmental monitoring, and selection of sample sites.
For example, QA establishes the overall contamination control strategy (CCS) that defines and assesses the effectiveness of all critical control points, including design, procedural, technical, and organizational controls employed to manage contamination risks. This strategy encompasses the entire facility and provides a comprehensive framework for contamination prevention.
Risk Management and Assessment
QA implements quality risk management principles to provide a proactive means of identifying, scientifically evaluating, and controlling potential risks to quality. This involves conducting thorough risk assessments that cover all human interactions with clean room areas, equipment placement and ergonomics, and air quality considerations. The risk-based approach ensures that monitoring efforts are focused on the most critical areas and processes where contamination could have the greatest impact on product quality.
QA also establishes risk-based environmental monitoring programs that are re-evaluated at defined intervals to confirm effectiveness, considering factors such as facility aging, barrier and cleanroom design optimization, and personnel changes. This ongoing assessment ensures that the monitoring program remains relevant and effective as conditions change over time.
Procedural Oversight and Documentation
QA ensures the development and maintenance of standardized operating procedures (SOPs) for all aspects of environmental monitoring, including air sampling, surface sampling, and personnel sampling protocols. These procedures ensure consistency in monitoring activities and provide clear guidance for personnel conducting environmental monitoring tasks.
The documentation responsibilities of QA extend to creating comprehensive quality management plans that clearly define responsibilities and duties to ensure that environmental monitoring data generated are of the required type, quality, and quantity. This includes establishing procedures for data analysis, trending, investigative responses to action level excursions, and appropriate corrective and preventative actions.
Compliance Assurance and Regulatory Alignment
QA ensures that environmental monitoring protocols meet Good Manufacturing Practice (GMP) requirements and align with current regulatory expectations such as the EU Annex 1 guidelines.
QA also manages the overall quality system to ensure that environmental monitoring activities support regulatory compliance and facilitate successful inspections and audits. This involves maintaining proper documentation, training records, and quality improvement processes that demonstrate ongoing commitment to contamination control.
QC Contributions to Environmental Monitoring and Contamination Control
Execution of Testing and Sampling
Quality Control is responsible for the hands-on execution of environmental monitoring testing protocols. QC personnel conduct microbiological testing including bioburden and endotoxin testing, as well as particle counting for non-viable particulate monitoring. This includes performing microbial air sampling using techniques such as active air sampling and settle plates, along with surface and personnel sampling using swabbing and contact plates.
For example, QC technicians perform routine environmental monitoring of classified manufacturing and filling areas, conducting both routine and investigational sampling to assess environmental conditions. They utilize calibrated active air samplers and strategically placed settle plates throughout cleanrooms, while also conducting surface and personnel sampling periodically, especially after critical interventions.
Data Analysis and Trend Monitoring
QC plays a crucial role in analyzing environmental monitoring data and identifying trends that may indicate potential contamination issues. When alert or action limits are exceeded, QC personnel initiate immediate investigations and document findings according to established protocols. This includes performing regular trend analysis on collected data to understand the state of control in cleanrooms and identify potential contamination risks before they lead to significant problems.
QC also maintains environmental monitoring programs and ensures all data is properly logged into Laboratory Information Management Systems (LIMS) for comprehensive tracking and analysis . This systematic approach to data management enables effective trending and supports decision-making processes related to contamination control.
Validation and Verification Activities
QC conducts critical validation activities to simulate aseptic processes and verify the effectiveness of contamination control measures. These activities provide direct evidence that manufacturing processes maintain sterility and/or bioburden control and that environmental controls are functioning as intended.
QC also performs specific testing protocols including dissolution testing, stability testing, and comprehensive analysis of finished products to ensure they meet quality specifications and are free from contamination. This testing provides the verification that QA-established systems are effectively preventing contamination.
Real-Time Monitoring and Response
QC supports continuous monitoring efforts through the implementation of Process Analytical Technology (PAT) for real-time quality verification. This includes continuous monitoring of non-viable particulates, which helps detect events that could potentially increase contamination risk and enables immediate corrective measures.
When deviations occur, QC personnel immediately report findings and place products on hold for further evaluation, providing documented reports and track-and-trend data to support decision-making processes. This rapid response capability is essential for preventing contaminated products from reaching the market.
Conclusion
While Quality Assurance and Quality Control in pharmaceutical manufacturing represent distinct processes with different focuses and approaches, they form a complementary continuum that ensures product quality throughout the lifecycle. QA is proactive, process-oriented, and focused on preventing quality issues through robust systems and procedures. QC is reactive, product-oriented, and focused on detecting and addressing quality issues through testing and inspection.
The organizational structure of quality functions in pharmaceutical companies varies, with models ranging from integrated quality units to separate departments, centralized or decentralized organizations, and matrix structures. Regardless of the organizational model, the Quality Unit plays a critical role in overseeing all quality-related activities and ensuring compliance with regulatory requirements.
The Pharmaceutical Quality System provides an overarching framework that integrates QA and QC activities within a comprehensive approach to quality management. By implementing effective quality systems and fostering a culture of quality, pharmaceutical companies can ensure the safety, efficacy, and quality of their products while meeting regulatory requirements and continuously improving their processes.
The pharmaceutical industry is navigating a transformative period in contamination control, driven by the convergence of updated international standards. The U.S. Pharmacopeia’s draft chapter〈1110〉 Microbial Contamination Control Strategy Considerations (March 2025) joins EU GMP Annex 1 (2022) in emphasizing risk-based strategies but differ in technical requirements and classification systems.
USP〈1110〉: A Lifecycle-Oriented Microbial Control Framework
The draft USP chapter introduces a comprehensive contamination control strategy (CCS) that spans the entire product lifecycle, from facility design to post-market surveillance. It emphasizes microbial, endotoxin, and pyrogen risks, requiring manufacturers to integrate quality risk management (QRM) into every operational phase. Facilities must adopt ISO 14644-1 cleanroom classifications, with ISO Class 5 (≤3,520 particles ≥0.5 µm/m³) mandated for aseptic processing areas. Environmental monitoring programs must include both viable (microbial) and nonviable particles, with data trends analyzed quarterly to refine alert/action levels. Unlike Annex 1, USP allows flexibility in risk assessment methodologies but mandates documented justifications for control measures, such as the use of closed systems or isolators to minimize human intervention.
EU GMP Annex 1: Granular Cleanroom and Sterilization Requirements
Annex 1 builds on ISO 14644-1 cleanroom standards but introduces pharmaceutical-specific adaptations through its Grade A–D system. Grade A zones (critical processing areas) require ISO Class 5 conditions during both “at-rest” and “in-operation” states, with continuous particle monitoring and microbial limits of <1 CFU/m³. Annex 1 also mandates smoke studies to validate unidirectional airflow patterns in Grade A areas, a requirement absent in ISO 14644-1. Sterilization processes, such as autoclaving and vaporized hydrogen peroxide (VHP) treatments, require pre- and post-use integrity testing, aligning with its focus on sterility assurance.
Reconciling Annex 1 and ISO 14644-1 Cleanroom Classifications
While both frameworks reference ISO 14644-1, Annex 1 overlays additional pharmaceutical requirements:
Aspect
EU GMP Annex 1
ISO 14644-1
Classification System
Grades A–D mapped to ISO classes
ISO Class 1–9 based on particle counts
Particle Size
≥0.5 µm and ≥5.0 µm monitoring for Grades A–B
≥0.1 µm to ≥5.0 µm, depending on class
Microbial Limits
Explicit CFU/m³ limits for each grade
No microbial criteria; focuses on particles
Operational States
Qualification required for “at-rest” and “in-operation” states
Single-state classification permitted
Airflow Validation
Smoke studies mandatory for Grade A
Airflow pattern testing optional
For example, a Grade B cleanroom (ISO Class 7 at rest) must maintain ISO Class 7 particle counts during production but adheres to stricter microbial limits (≤10 CFU/m³) than ISO 14644-1 alone. Manufacturers must design monitoring programs that satisfy both standards, such as deploying continuous particle counters for Annex 1 compliance while maintaining ISO certification reports.
Classification
Description
Grade A
Critical area for high-risk and aseptic operations that corresponds to ISO 5 at rest/static and ISO 4.8 (in-operation/dynamic). Grade A areas apply to aseptic operations where the sterile product, product primary packaging components and product-contact surfaces are exposed to the environment. Normally Grade A conditions are provided by localized air flow protection, such as unidirectional airflow workstations within a Restricted Access Barrier System (RABS) or isolator. Direct intervention (e.g., without the protection of barrier and glove port protection) into the Grade A area by operators must be minimized by premises, equipment, process, or procedural design.
Grade B
For aseptic preparation and filling, this is the background area for Grade A (where it is not an isolator) and corresponds to ISO 5 at rest/static and ISO 7 in-operation/dynamic. Air pressure differences must be continuously monitored. Classified spaces of lower grade can be considered with the appropriate risk assessment and technical justification.
Grade C
Used for carrying out less critical steps in the manufacture of aseptically filled sterile products or as a background for isolators. They can also be used for the preparation/filling of terminally sterilized products. Grade C correspond to ISO 7 at rest/static and ISO 8 in-operation/dynamic.
Grade D
Used to carry out non-sterile operations and corresponds to ISO 8 at rest/static and in-operation/dynamic.
Both frameworks require Quality Risk Management. USP〈1110〉advocates for a flexible, science-driven approach, allowing tools like HACCP (Hazard Analysis Critical Control Points) or FMEA (Failure Modes Effects Analysis) to identify critical control points. For instance, a biologics manufacturer might use HACCP to prioritize endotoxin controls during cell culture harvesting. USP also emphasizes lifecycle risk reviews, requiring CCS updates after facility modifications or adverse trend detections.
Annex 1 mandates formal QRM processes with documented risk assessments for all sterilization and aseptic processes. Its Annex 1.25 clause requires FMEA for media fill simulations, ensuring worst-case scenarios (e.g., maximum personnel presence) are tested. Risk assessments must also justify cleanroom recovery times after interventions, linking airflow validation data to contamination probability.
A harmonized approach involves:
Baseline Risk Identification: Use HACCP to map contamination risks across product stages, aligning with USP’s lifecycle focus.
Control Measure Integration: Apply Annex 1’s sterilization and airflow requirements to critical risks identified in USP’s CCS.
Continuous Monitoring: Combine USP’s trend analysis with continuous monitoring for real-time risk mitigation.
Strategic Implementation Considerations
Reconciling these standards requires a multi-layered strategy. Facilities must first achieve ISO 14644-1 certification for particle counts, then overlay Annex 1’s microbial and operational requirements. For example, an ISO Class 7 cleanroom used for vial filling would need Grade B microbial monitoring (≤10 CFU/m³) and quarterly smoke studies to validate airflow. Risk management documentation should cross-reference USP’s CCS objectives with Annex 1’s sterilization validations, creating a unified audit trail. Training programs must blend USP’s aseptic technique modules with Annex 1’s cleanroom behavior protocols, ensuring personnel understand both particle control and microbial hygiene.
Toward Global Harmonization
The draft USP〈1110〉and Annex 1 represent complementary pillars of modern contamination control. By anchoring cleanroom designs to ISO 14644-1 and layering region-specific requirements, manufacturers can streamline compliance across jurisdictions. Proactive risk management—combining USP’s flexibility with Annex 1’s rigor—will be pivotal in navigating this evolving landscape. As regulatory expectations converge, firms that invest in integrated CCS platforms will gain agility in an increasingly complex global market.
In a past post discussing the program level in the document hierarchy, I outlined how program documents serve as critical connective tissue between high-level policies and detailed procedures. Today, I’ll explore three distinct but related approaches to control strategies: the Annex 1 Contamination Control Strategy (CCS), the ICH Q8 Process Control Strategy, and a Technology Platform Control Strategy. Understanding their differences and relationships allows us to establish a comprehensive quality system in pharmaceutical manufacturing, especially as regulatory requirements continue to evolve and emphasize more scientific, risk-based approaches to quality management.
Control strategies have evolved significantly and are increasingly central to pharmaceutical quality management. As I noted in my previous article, program documents create an essential mapping between requirements and execution, demonstrating the design thinking that underpins our quality processes. Control strategies exemplify this concept, providing comprehensive frameworks that ensure consistent product quality through scientific understanding and risk management.
The pharmaceutical industry has gradually shifted from reactive quality testing to proactive quality design. This evolution mirrors the maturation of our document hierarchies, with control strategies occupying that critical program-level space between overarching quality policies and detailed operational procedures. They serve as the blueprint for how quality will be achieved, maintained, and improved throughout a product’s lifecycle.
This evolution has been accelerated by increasing regulatory scrutiny, particularly following numerous drug recalls and contamination events resulting in significant financial losses for pharmaceutical companies.
Annex 1 Contamination Control Strategy: A Facility-Focused Approach
The Annex 1 Contamination Control Strategy represents a comprehensive, facility-focused approach to preventing chemical, physical and microbial contamination in pharmaceutical manufacturing environments. The CCS takes a holistic view of the entire manufacturing facility rather than focusing on individual products or processes.
A properly implemented CCS requires a dedicated cross-functional team representing technical knowledge from production, engineering, maintenance, quality control, microbiology, and quality assurance. This team must systematically identify contamination risks throughout the facility, develop mitigating controls, and establish monitoring systems that provide early detection of potential issues. The CCS must be scientifically formulated and tailored specifically for each manufacturing facility’s unique characteristics and risks.
What distinguishes the Annex 1 CCS is its infrastructural approach to Quality Risk Management. Rather than focusing solely on product attributes or process parameters, it examines how facility design, environmental controls, personnel practices, material flow, and equipment operate collectively to prevent contamination. The CCS process involves continual identification, scientific evaluation, and effective control of potential contamination risks to product quality.
Critical Factors in Developing an Annex 1 CCS
The development of an effective CCS involves several critical considerations. According to industry experts, these include identifying the specific types of contaminants that pose a risk, implementing appropriate detection methods, and comprehensively understanding the potential sources of contamination. Additionally, evaluating the risk of contamination and developing effective strategies to control and minimize such risks are indispensable components of an efficient contamination control system.
When implementing a CCS, facilities should first determine their critical control points. Annex 1 highlights the importance of considering both plant design and processes when developing a CCS. The strategy should incorporate a monitoring and ongoing review system to identify potential lapses in the aseptic environment and contamination points in the facility. This continuous assessment approach ensures that contamination risks are promptly identified and addressed before they impact product quality.
ICH Q8 Process Control Strategy: The Quality by Design Paradigm
While the Annex 1 CCS focuses on facility-wide contamination prevention, the ICH Q8 Process Control Strategy takes a product-centric approach rooted in Quality by Design (QbD) principles. The ICH Q8(R2) guideline introduces control strategy as “a planned set of controls derived from current product and process understanding that ensures process performance and product quality”. This approach emphasizes designing quality into products rather than relying on final testing to detect issues.
The ICH Q8 guideline outlines a set of key principles that form the foundation of an effective process control strategy. At its core is pharmaceutical development, which involves a comprehensive understanding of the product and its manufacturing process, along with identifying critical quality attributes (CQAs) that impact product safety and efficacy. Risk assessment plays a crucial role in prioritizing efforts and resources to address potential issues that could affect product quality.
The development of an ICH Q8 control strategy follows a systematic sequence: defining the Quality Target Product Profile (QTPP), identifying Critical Quality Attributes (CQAs), determining Critical Process Parameters (CPPs) and Critical Material Attributes (CMAs), and establishing appropriate control methods. This scientific framework enables manufacturers to understand how material attributes and process parameters affect product quality, allowing for more informed decision-making and process optimization.
Design Space and Lifecycle Approach
A unique aspect of the ICH Q8 control strategy is the concept of “design space,” which represents a range of process parameters within which the product will consistently meet desired quality attributes. Developing and demonstrating a design space provides flexibility in manufacturing without compromising product quality. This approach allows manufacturers to make adjustments within the established parameters without triggering regulatory review, thus enabling continuous improvement while maintaining compliance.
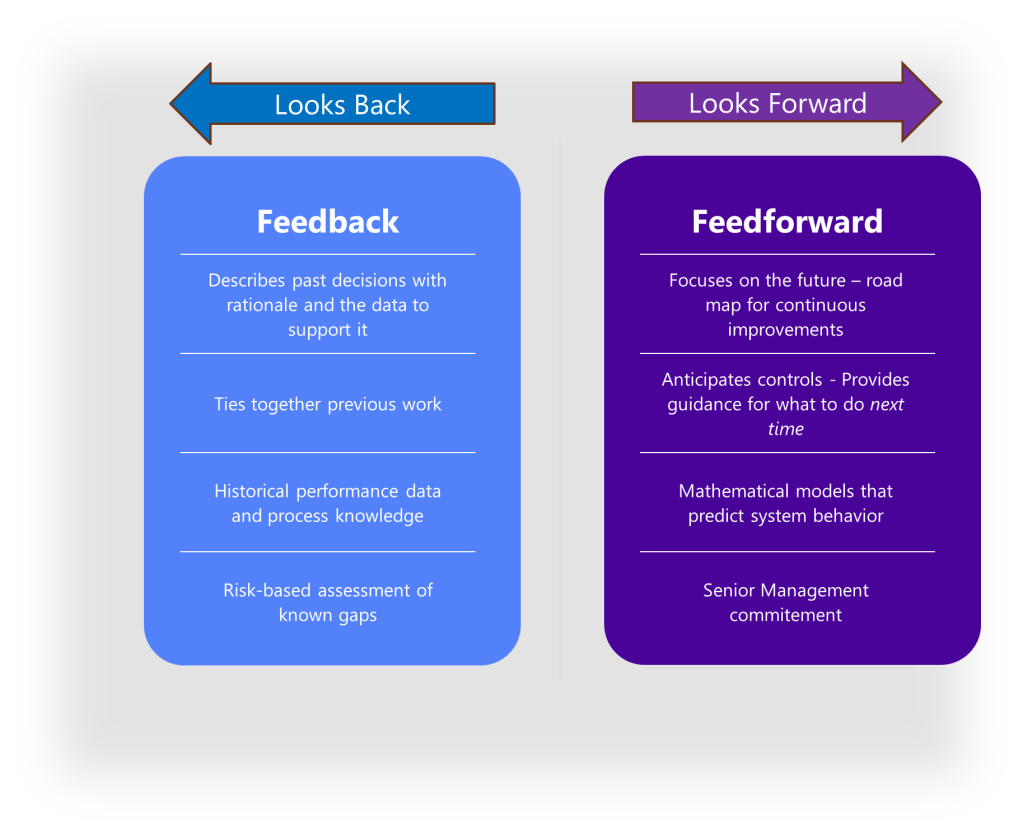
What makes the ICH Q8 control strategy distinct is its dynamic, lifecycle-oriented nature. The guideline encourages a lifecycle approach to product development and manufacturing, where continuous improvement and monitoring are carried out throughout the product’s lifecycle, from development to post-approval. This approach creates a feedback-feedforward “controls hub” that integrates risk management, knowledge management, and continuous improvement throughout the product lifecycle.
Technology Platform Control Strategies: Leveraging Prior Knowledge
As pharmaceutical development becomes increasingly complex, particularly in emerging fields like cell and gene therapies, technology platform control strategies offer an approach that leverages prior knowledge and standardized processes to accelerate development while maintaining quality standards. Unlike product-specific control strategies, platform strategies establish common processes, parameters, and controls that can be applied across multiple products sharing similar characteristics or manufacturing approaches.
The importance of maintaining state-of-the-art technology platforms has been highlighted in recent regulatory actions. A January 2025 FDA Warning Letter to Sanofi, concerning a facility that had previously won the ISPE’s Facility of the Year award in 2020, emphasized the requirement for “timely technological upgrades to equipment/facility infrastructure”. This regulatory focus underscores that even relatively new facilities must continually evolve their technological capabilities to maintain compliance and product quality.
Developing a Comprehensive Technology Platform Roadmap
A robust technology platform control strategy requires a well-structured technology roadmap that anticipates both regulatory expectations and technological advancements. According to recent industry guidance, this roadmap should include several key components:
At its foundation, regular assessment protocols are essential. Organizations should conduct comprehensive annual evaluations of platform technologies, examining equipment performance metrics, deviations associated with the platform, and emerging industry standards that might necessitate upgrades. These assessments should be integrated with Facility and Utility Systems Effectiveness (FUSE) metrics and evaluated through structured quality governance processes.
The technology roadmap must also incorporate systematic methods for monitoring industry trends. This external vigilance ensures platform technologies remain current with evolving expectations and capabilities.
Risk-based prioritization forms another critical element of the platform roadmap. By utilizing living risk assessments, organizations can identify emerging issues and prioritize platform upgrades based on their potential impact on product quality and patient safety. These assessments should represent the evolution of the original risk management that established the platform, creating a continuous thread of risk evaluation throughout the platform’s lifecycle.
Implementation and Verification of Platform Technologies
Successful implementation of platform technologies requires robust change management procedures. These should include detailed documentation of proposed platform modifications, impact assessments on product quality across the portfolio, appropriate verification activities, and comprehensive training programs. This structured approach ensures that platform changes are implemented systematically with full consideration of their potential implications.
Verification activities for platform technologies must be particularly thorough, given their application across multiple products. The commissioning, qualification, and validation activities should demonstrate not only that platform components meet predetermined specifications but also that they maintain their intended performance across the range of products they support. This verification must consider the variability in product-specific requirements while confirming the platform’s core capabilities.
Continuous monitoring represents the final essential element of platform control strategies. By implementing ongoing verification protocols aligned with Stage 3 of the FDA’s process validation model, organizations can ensure that platform technologies remain in a state of control during routine commercial manufacture. This monitoring should anticipate and prevent issues, detect unplanned deviations, and identify opportunities for platform optimization.
Leveraging Advanced Technologies in Platform Strategies
Modern technology platforms increasingly incorporate advanced capabilities that enhance their flexibility and performance. Single-Use Systems (SUS) reduce cleaning and validation requirements while improving platform adaptability across products. Modern Microbial Methods (MMM) offer advantages over traditional culture-based approaches in monitoring platform performance. Process Analytical Technology (PAT) enables real-time monitoring and control, enhancing product quality and process understanding across the platform. Data analytics and artificial intelligence tools identify trends, predict maintenance needs, and optimize processes across the product portfolio.
The implementation of these advanced technologies within platform strategies creates significant opportunities for standardization, knowledge transfer, and continuous improvement. By establishing common technological foundations that can be applied across multiple products, organizations can accelerate development timelines, reduce validation burdens, and focus resources on understanding the unique aspects of each product while maintaining a robust quality foundation.
How Control Strategies Tie Together Design, Qualification/Validation, and Risk Management
Control strategies serve as the central nexus connecting design, qualification/validation, and risk management in a comprehensive quality framework. This integration is not merely beneficial but essential for ensuring product quality while optimizing resources. A well-structured control strategy creates a coherent narrative from initial concept through on-going production, ensuring that design intentions are preserved through qualification activities and ongoing risk management.
During the design phase, scientific understanding of product and process informs the development of the control strategy. This strategy then guides what must be qualified and validated and to what extent. Rather than validating everything (which adds cost without necessarily improving quality), the control strategy directs validation resources toward aspects most critical to product quality.
The relationship works in both directions—design decisions influence what will require validation, while validation capabilities and constraints may inform design choices. For example, a process designed with robust, well-understood parameters may require less extensive validation than one operating at the edge of its performance envelope. The control strategy documents this relationship, providing scientific justification for validation decisions based on product and process understanding.
Risk management principles are foundational to modern control strategies, informing both design decisions and priorities. A systematic risk assessment approach helps identify which aspects of a process or facility pose the greatest potential impact on product quality and patient safety. The control strategy then incorporates appropriate controls and monitoring systems for these high-risk elements, ensuring that validation efforts are proportionate to risk levels.
The Feedback-Feedforward Mechanism
One of the most powerful aspects of an integrated control strategy is its ability to function as what experts call a feedback-feedforward controls hub. As a product moves through its lifecycle, from development to commercial manufacturing, the control strategy evolves based on accumulated knowledge and experience. Validation results, process monitoring data, and emerging risks all feed back into the control strategy, which in turn drives adjustments to design parameters and validation approaches.
Comparing Control Strategy Approaches: Similarities and Distinctions
While these three control strategy approaches have distinct focuses and applications, they share important commonalities. All three emphasize scientific understanding, risk management, and continuous improvement. They all serve as program-level documents that connect high-level requirements with operational execution. And all three have gained increasing regulatory recognition as pharmaceutical quality management has evolved toward more systematic, science-based approaches.
Aspect
Annex 1 CCS
ICH Q8 Process Control Strategy
Technology Platform Control Strategy
Primary Focus
Facility-wide contamination prevention
Product and process quality
Standardized approach across multiple products
Scope
Microbial, pyrogen, and particulate contamination (a good one will focus on physical, chemical and biologic hazards)
All aspects of product quality
Common technology elements shared across products
Regulatory Foundation
EU GMP Annex 1 (2022 revision)
ICH Q8(R2)
Emerging FDA guidance (Platform Technology Designation)
Implementation Level
Manufacturing facility
Individual product
Technology group or platform
Key Components
Contamination risk identification, detection methods, understanding of contamination sources
QTPP, CQAs, CPPs, CMAs, design space
Standardized technologies, processes, and controls
Risk Management Approach
Infrastructural (facility design, processes, personnel) – great for a HACCP
Product-specific (process parameters, material attributes)
Process analytical technology, real-time release testing
Platform data management and cross-product analytics
These approaches are not mutually exclusive; rather, they complement each other within a comprehensive quality management system. A manufacturing site producing sterile products needs both an Annex 1 CCS for facility-wide contamination control and ICH Q8 process control strategies for each product. If the site uses common technology platforms across multiple products, platform control strategies would provide additional efficiency and standardization.
Control Strategies Through the Lens of Knowledge Management: Enhancing Quality and Operational Excellence
The pharmaceutical industry’s approach to control strategies has evolved significantly in recent years, with systematic knowledge management emerging as a critical foundation for their effectiveness. Control strategies—whether focused on contamination prevention, process control, or platform technologies—fundamentally depend on how knowledge is created, captured, disseminated, and applied across an organization. Understanding the intersection between control strategies and knowledge management provides powerful insights into building more robust pharmaceutical quality systems and achieving higher levels of operational excellence.
The Knowledge Foundation of Modern Control Strategies
Control strategies represent systematic approaches to ensuring consistent pharmaceutical quality by managing various aspects of production. While these strategies differ in focus and application, they share a common foundation in knowledge—both explicit (documented) and tacit (experiential).
Knowledge Management as the Binding Element
The ICH Q10 Pharmaceutical Quality System model positions knowledge management alongside quality risk management as dual enablers of pharmaceutical quality. This pairing is particularly significant when considering control strategies, as it establishes what might be called a “Risk-Knowledge Infinity Cycle”—a continuous process where increased knowledge leads to decreased uncertainty and therefore decreased risk. Control strategies represent the formal mechanisms through which this cycle is operationalized in pharmaceutical manufacturing.
Effective control strategies require comprehensive knowledge visibility across functional areas and lifecycle phases. Organizations that fail to manage knowledge effectively often experience problems like knowledge silos, repeated issues due to lessons not learned, and difficulty accessing expertise or historical product knowledge—all of which directly impact the effectiveness of control strategies and ultimately product quality.
The Feedback-Feedforward Controls Hub: A Knowledge Integration Framework
As described above, the heart of effective control strategies lies is the “feedback-feedforward controls hub.” This concept represents the integration point where knowledge flows bidirectionally to continuously refine and improve control mechanisms. In this model, control strategies function not as static documents but as dynamic knowledge systems that evolve through continuous learning and application.
The feedback component captures real-time process data, deviations, and outcomes that generate new knowledge about product and process performance. The feedforward component takes this accumulated knowledge and applies it proactively to prevent issues before they occur. This integrated approach creates a self-reinforcing cycle where control strategies become increasingly sophisticated and effective over time.
For example, in an ICH Q8 process control strategy, process monitoring data feeds back into the system, generating new understanding about process variability and performance. This knowledge then feeds forward to inform adjustments to control parameters, risk assessments, and even design space modifications. The hub serves as the central coordination mechanism ensuring these knowledge flows are systematically captured and applied.
Knowledge Flow Within Control Strategy Implementation
Knowledge flows within control strategies typically follow the knowledge management process model described in the ISPE Guide, encompassing knowledge creation, curation, dissemination, and application. For control strategies to function effectively, this flow must be seamless and well-governed.
The systematic management of knowledge within control strategies requires:
Methodical capture of knowledge through various means appropriate to the control strategy context
Proper identification, review, and analysis of this knowledge to generate insights
Effective storage and visibility to ensure accessibility across the organization
Clear pathways for knowledge application, transfer, and growth
When these elements are properly integrated, control strategies benefit from continuous knowledge enrichment, resulting in more refined and effective controls. Conversely, barriers to knowledge flow—such as departmental silos, system incompatibilities, or cultural resistance to knowledge sharing—directly undermine the effectiveness of control strategies.
Annex 1 Contamination Control Strategy Through a Knowledge Management Lens
The Annex 1 Contamination Control Strategy represents a facility-focused approach to preventing microbial, pyrogen, and particulate contamination. When viewed through a knowledge management lens, the CCS becomes more than a compliance document—it emerges as a comprehensive knowledge system integrating multiple knowledge domains.
Effective implementation of an Annex 1 CCS requires managing diverse knowledge types across functional boundaries. This includes explicit knowledge documented in environmental monitoring data, facility design specifications, and cleaning validation reports. Equally important is tacit knowledge held by personnel about contamination risks, interventions, and facility-specific nuances that are rarely fully documented.
The knowledge management challenges specific to contamination control include ensuring comprehensive capture of contamination events, facilitating cross-functional knowledge sharing about contamination risks, and enabling access to historical contamination data and prior knowledge. Organizations that approach CCS development with strong knowledge management practices can create living documents that continuously evolve based on accumulated knowledge rather than static compliance tools.
Knowledge mapping is particularly valuable for CCS implementation, helping to identify critical contamination knowledge sources and potential knowledge gaps. Communities of practice spanning quality, manufacturing, and engineering functions can foster collaboration and tacit knowledge sharing about contamination control. Lessons learned processes ensure that insights from contamination events contribute to continuous improvement of the control strategy.
ICH Q8 Process Control Strategy: Quality by Design and Knowledge Management
The ICH Q8 Process Control Strategy embodies the Quality by Design paradigm, where product and process understanding drives the development of controls that ensure consistent quality. This approach is fundamentally knowledge-driven, making effective knowledge management essential to its success.
The QbD approach begins with applying prior knowledge to establish the Quality Target Product Profile (QTPP) and identify Critical Quality Attributes (CQAs). Experimental studies then generate new knowledge about how material attributes and process parameters affect these quality attributes, leading to the definition of a design space and control strategy. This sequence represents a classic knowledge creation and application cycle that must be systematically managed.
Knowledge management challenges specific to ICH Q8 process control strategies include capturing the scientific rationale behind design choices, maintaining the connectivity between risk assessments and control parameters, and ensuring knowledge flows across development and manufacturing boundaries. Organizations that excel at knowledge management can implement more robust process control strategies by ensuring comprehensive knowledge visibility and application.
Particularly important for process control strategies is the management of decision rationale—the often-tacit knowledge explaining why certain parameters were selected or why specific control approaches were chosen. Explicit documentation of this decision rationale ensures that future changes to the process can be evaluated with full understanding of the original design intent, avoiding unintended consequences.
Technology Platform Control Strategies: Leveraging Knowledge Across Products
Technology platform control strategies represent standardized approaches applied across multiple products sharing similar characteristics or manufacturing technologies. From a knowledge management perspective, these strategies exemplify the power of knowledge reuse and transfer across product boundaries.
The fundamental premise of platform approaches is that knowledge gained from one product can inform the development and control of similar products, creating efficiencies and reducing risks. This depends on robust knowledge management practices that make platform knowledge visible and available across product teams and lifecycle phases.
Knowledge management challenges specific to platform control strategies include ensuring consistent knowledge capture across products, facilitating cross-product learning, and balancing standardization with product-specific requirements. Organizations with mature knowledge management practices can implement more effective platform strategies by creating knowledge repositories, communities of practice, and lessons learned processes that span product boundaries.
Integrating Control Strategies with Design, Qualification/Validation, and Risk Management
Control strategies serve as the central nexus connecting design, qualification/validation, and risk management in a comprehensive quality framework. This integration is not merely beneficial but essential for ensuring product quality while optimizing resources. A well-structured control strategy creates a coherent narrative from initial concept through commercial production, ensuring that design intentions are preserved through qualification activities and ongoing risk management.
The Design-Validation Continuum
Control strategies form a critical bridge between product/process design and validation activities. During the design phase, scientific understanding of the product and process informs the development of the control strategy. This strategy then guides what must be validated and to what extent. Rather than validating everything (which adds cost without necessarily improving quality), the control strategy directs validation resources toward aspects most critical to product quality.
The relationship works in both directions—design decisions influence what will require validation, while validation capabilities and constraints may inform design choices. For example, a process designed with robust, well-understood parameters may require less extensive validation than one operating at the edge of its performance envelope. The control strategy documents this relationship, providing scientific justification for validation decisions based on product and process understanding.
Risk-Based Prioritization
Risk management principles are foundational to modern control strategies, informing both design decisions and validation priorities. A systematic risk assessment approach helps identify which aspects of a process or facility pose the greatest potential impact on product quality and patient safety. The control strategy then incorporates appropriate controls and monitoring systems for these high-risk elements, ensuring that validation efforts are proportionate to risk levels.
The Feedback-Feedforward Mechanism
The feedback-feedforward controls hub represents a sophisticated integration of two fundamental control approaches, creating a central mechanism that leverages both reactive and proactive control strategies to optimize process performance. This concept emerges as a crucial element in modern control systems, particularly in pharmaceutical manufacturing, chemical processing, and advanced mechanical systems.
To fully grasp the concept of a feedback-feedforward controls hub, we must first distinguish between its two primary components. Feedback control works on the principle of information from the outlet of a process being “fed back” to the input for corrective action. This creates a loop structure where the system reacts to deviations after they occur. Fundamentally reactive in nature, feedback control takes action only after detecting a deviation between the process variable and setpoint.
In contrast, feedforward control operates on the principle of preemptive action. It monitors load variables (disturbances) that affect a process and takes corrective action before these disturbances can impact the process variable. Rather than waiting for errors to manifest, feedforward control uses data from load sensors to predict when an upset is about to occur, then feeds that information forward to the final control element to counteract the load change proactively.
The feedback-feedforward controls hub serves as a central coordination point where these two control strategies converge and complement each other. As a product moves through its lifecycle, from development to commercial manufacturing, this control hub evolves based on accumulated knowledge and experience. Validation results, process monitoring data, and emerging risks all feed back into the control strategy, which in turn drives adjustments to design parameters and validation approaches.
Knowledge Management Maturity in Control Strategy Implementation
The effectiveness of control strategies is directly linked to an organization’s knowledge management maturity. Organizations with higher knowledge management maturity typically implement more robust, science-based control strategies that evolve effectively over time. Conversely, organizations with lower maturity often struggle with static control strategies that fail to incorporate learning and experience.
Common knowledge management gaps affecting control strategies include:
Inadequate mechanisms for capturing tacit knowledge from subject matter experts
Poor visibility of knowledge across organizational and lifecycle boundaries
Ineffective lessons learned processes that fail to incorporate insights into control strategies
Limited knowledge sharing between sites implementing similar control strategies
Difficulty accessing historical knowledge that informed original control strategy design
Addressing these gaps through systematic knowledge management practices can significantly enhance control strategy effectiveness, leading to more robust processes, fewer deviations, and more efficient responses to change.
The examination of control strategies through a knowledge management lens reveals their fundamentally knowledge-dependent nature. Whether focused on contamination control, process parameters, or platform technologies, control strategies represent the formal mechanisms through which organizational knowledge is applied to ensure consistent pharmaceutical quality.
Organizations seeking to enhance their control strategy effectiveness should consider several key knowledge management principles:
Recognize both explicit and tacit knowledge as essential components of effective control strategies
Ensure knowledge flows seamlessly across functional boundaries and lifecycle phases
Address all four pillars of knowledge management—people, process, technology, and governance
Implement systematic methods for capturing lessons and insights that can enhance control strategies
Foster a knowledge-sharing culture that supports continuous learning and improvement
By integrating these principles into control strategy development and implementation, organizations can create more robust, science-based approaches that continuously evolve based on accumulated knowledge and experience. This not only enhances regulatory compliance but also improves operational efficiency and product quality, ultimately benefiting patients through more consistent, high-quality pharmaceutical products.
The feedback-feedforward controls hub concept represents a particularly powerful framework for thinking about control strategies, emphasizing the dynamic, knowledge-driven nature of effective controls. By systematically capturing insights from process performance and proactively applying this knowledge to prevent issues, organizations can create truly learning control systems that become increasingly effective over time.
Conclusion: The Central Role of Control Strategies in Pharmaceutical Quality Management
Control strategies—whether focused on contamination prevention, process control, or technology platforms—serve as the intellectual foundation connecting high-level quality policies with detailed operational procedures. They embody scientific understanding, risk management decisions, and continuous improvement mechanisms in a coherent framework that ensures consistent product quality.
Regulatory Needs and Control Strategies
Regulatory guidelines like ICH Q8 and Annex 1 CCS underscore the importance of control strategies in ensuring product quality and compliance. ICH Q8 emphasizes a Quality by Design (QbD) approach, where product and process understanding drives the development of controls. Annex 1 CCS focuses on facility-wide contamination prevention, highlighting the need for comprehensive risk management and control systems. These regulatory expectations necessitate robust control strategies that integrate scientific knowledge with operational practices.
Knowledge Management: The Backbone of Effective Control Strategies
Knowledge management (KM) plays a pivotal role in the effectiveness of control strategies. By systematically acquiring, analyzing, storing, and disseminating information related to products and processes, organizations can ensure that the right knowledge is available at the right time. This enables informed decision-making, reduces uncertainty, and ultimately decreases risk.
Risk Management and Control Strategies
Risk management is inextricably linked with control strategies. By identifying and mitigating risks, organizations can maintain a state of control and facilitate continual improvement. Control strategies must be designed to incorporate risk assessments and management processes, ensuring that they are proactive and adaptive.
The Interconnectedness of Control Strategies
Control strategies are not isolated entities but are interconnected with design, qualification/validation, and risk management processes. They form a feedback-feedforward controls hub that evolves over a product’s lifecycle, incorporating new insights and adjustments based on accumulated knowledge and experience. This dynamic approach ensures that control strategies remain effective and relevant, supporting both regulatory compliance and operational excellence.
Why Control Strategies Are Key
Control strategies are essential for several reasons:
Regulatory Compliance: They ensure adherence to regulatory guidelines and standards, such as ICH Q8 and Annex 1 CCS.
Quality Assurance: By integrating scientific understanding and risk management, control strategies guarantee consistent product quality.
Operational Efficiency: Effective control strategies streamline processes, reduce waste, and enhance productivity.
Knowledge Management: They facilitate the systematic management of knowledge, ensuring that insights are captured and applied across the organization.
Risk Mitigation: Control strategies proactively identify and mitigate risks, protecting both product quality and patient safety.
Control strategies represent the central mechanism through which pharmaceutical companies ensure quality, manage risk, and leverage knowledge. As the industry continues to evolve with new technologies and regulatory expectations, the importance of robust, science-based control strategies will only grow. By integrating knowledge management, risk management, and regulatory compliance, organizations can develop comprehensive quality systems that protect patients, satisfy regulators, and drive operational excellence.
The pharmaceutical industry stands at an inflection point in microbial control, with bacterial endotoxin management undergoing a profound transformation. For decades, compliance focused on meeting pharmacopeial limits at product release—notably the 5.0 EU/kg threshold for parenterals mandated by standards like Ph. Eur. 5.1.10. While these endotoxin specifications remain enshrined as Critical Quality Attributes (CQAs), regulators now demand a fundamental reimagining of control strategies that transcends product specifications.
This shift reflects growing recognition that endotoxin contamination is fundamentally a facility-driven risk rather than a product-specific property. Health Authorities increasingly expect manufacturers to implement preventive, facility-wide control strategies anchored in quantitative risk modeling, rather than relying on end-product testing.
The EU Annex 1 Contamination Control Strategy (CCS) framework crystallizes this evolution, requiring cross-functional systems that integrate:
Process design capable of achieving ≥3 log10 endotoxin reduction (LRV) with statistical confidence (p<0.01)
Real-time monitoring of critical utilities like WFI and clean steam
Personnel flow controls to minimize bioburden ingress
Our organizations should be working to bridge the gap between compendial compliance and true contamination control—from implementing predictive analytics for endotoxin risk scoring to designing closed processing systems with inherent contamination barriers. We’ll examine why traditional quality-by-testing approaches are yielding to facility-driven quality-by-design strategies, and how leading organizations are leveraging computational fluid dynamics and risk-based control charts to stay ahead of regulatory expectations.
Bacterial Endotoxins: Bridging Compendial Safety and Facility-Specific Risks
Bacterial endotoxins pose unique challenges as their control depends on facility infrastructure rather than process parameters alone. Unlike sterility assurance, which can be validated through autoclave cycles, endotoxin control requires continuous vigilance over water systems, HVAC performance, and material sourcing. The compendial limit of 5.0 EU/kg ensures pyrogen-free products, but HAs argue this threshold does not account for facility-wide contamination risks that could compromise multiple batches. For example, a 2023 EMA review found 62% of endotoxin-related recalls stemmed from biofilm breaches in water-for-injection (WFI) systems rather than product-specific failures.
Annex 1 addresses this through CCS requirements that mandate:
Tiered control limits integrating compendial safety thresholds (specifications) with preventive action limits (in-process controls)
Lifecycle validation of sterilization processes, hold times, and monitoring systems
Annex 1’s Contamination Control Strategy: A Blueprint for Endotoxin Mitigation
Per Annex 1’s glossary, a CCS is “a planned set of controls […] derived from product and process understanding that assures process performance and product quality”. For endotoxins, this translates to 16 interrelated elements outlined in Annex 1’s Section 2.6, including:
The revised Annex 1 mandates Quality Risk Management (QRM) per ICH Q9, requiring facilities to deploy appropriate risk management.
Hazard Analysis and Critical Control Points (HACCP) identifies critical control points (CCPs) where endotoxin ingress or proliferation could occur. For there a Failure Modes Effects and Criticality Analysis (FMECA) can further prioritizes risks based on severity, occurrence, and detectability.
Endotoxin-Specific FMECA (Failure Mode, Effects, and Criticality Analysis)
Failure Mode
Severity (S)
Occurrence (O)
Detectability (D)
RPN (S×O×D)
Mitigation
WFI biofilm formation
5 (Product recall)
3 (1/2 years)
2 (Inline sensors)
30
Install ozone-resistant diaphragm valves
HVAC filter leakage
4 (Grade C contamination)
2 (1/5 years)
4 (Weekly integrity tests)
32
HEPA filter replacement every 6 months
Simplified FMECA for endotoxin control (RPN thresholds: <15=Low, 15-50=Medium, >50=High)
Process Validation and Analytical Controls
As outlined in the FDA’s Process Validation: General Principles and Practices, PV is structured into three stages: process design, process qualification, and continued process verification (CPV). For bacterial endotoxin control, PV extends to validating sterilization processes, hold times, and water-for-injection (WFI) systems, where CPPs like sanitization frequency and turbulent flow rates are tightly controlled to prevent biofilm formation.
Analytical controls form the backbone of quality assurance, with method validation per ICH Q2(R1) ensuring accuracy, precision, and specificity for critical tests such as endotoxin quantification. The advent of rapid microbiological methods (RMM), including recombinant Factor C (rFC) assays, has reduced endotoxin testing timelines from hours to minutes, enabling near-real-time release of drug substances. These methods are integrated into continuous process verification programs, where action limits—set at 50% of the assay’s limit of quantitation (LOQ)—serve as early indicators of facility-wide contamination risks. For example, inline sensors in WFI systems or bioreactors provide continuous endotoxin data, which is trended alongside environmental monitoring results to preempt deviations. The USP <1220> lifecycle approach further mandates ongoing method performance verification, ensuring analytical procedures adapt to process changes or scale-up.
The integration of Process Analytical Technology (PAT) and Quality by Design (QbD) principles has transformed manufacturing by embedding real-time quality controls into the process itself. PAT tools such as Raman spectroscopy and centrifugal microfluidics enable on-line monitoring of product titers and impurity profiles, while multivariate data analysis (MVDA) correlates CPPs with CQAs to refine design spaces. Regulatory submissions now emphasize integrated control strategies that combine process validation data, analytical lifecycle management, and facility-wide contamination controls—aligning with EU GMP Annex 1’s mandate for holistic contamination control strategies (CCS). By harmonizing PV with advanced analytics, manufacturers can navigate HA expectations for tighter in-process limits while ensuring patient safety through compendial-aligned specifications.
Single-use sensor networks: RFID-enabled endotoxin probes providing real-time CCS data
Advanced water system designs: Reverse osmosis (RO) and electrodeionization (EDI) systems with ≤0.001 EU/mL capability without distillation
Manufacturers can prioritize transforming endotoxin control from a compliance exercise into a strategic quality differentiator—ensuring patient safety while meeting HA expectations for preventive contamination management.