In my recent exploration of the Jobs-to-Be-Done tool I examined how customer-centric thinking could revolutionize our understanding of complex quality processes. Today, I want to extend that analysis to one of the most persistent challenges in pharmaceutical data integrity: determining when electronic signatures are truly required to meet regulatory standards and data integrity expectations.
Most organizations approach electronic signature decisions through what I call “compliance theater”—mechanically applying rules without understanding the fundamental jobs these signatures need to accomplish. They focus on regulatory checkbox completion rather than building genuine data integrity capability. This approach creates elaborate signature workflows that satisfy auditors but fail to serve the actual needs of users, processes, or the data integrity principles they’re meant to protect.
The cost of getting this wrong extends far beyond regulatory findings. When organizations implement electronic signatures incorrectly, they create false confidence in their data integrity controls while potentially undermining the very protections these signatures are meant to provide. Conversely, when they avoid electronic signatures where they would genuinely improve data integrity, they perpetuate manual processes that introduce unnecessary risks and inefficiencies.
The Electronic Signature Jobs Users Actually Hire
When quality professionals, process owners and system owners consider electronic signature requirements, what job are they really trying to accomplish? The answer reveals a profound disconnect between regulatory intent and operational reality.
The Core Functional Job
“When I need to ensure data integrity, establish accountability, and meet regulatory requirements for record authentication, I want a signature method that reliably links identity to action and preserves that linkage throughout the record lifecycle, so I can demonstrate compliance and maintain trust in my data.”
This job statement immediately exposes the inadequacy of most electronic signature decisions. Organizations often focus on technical implementation rather than the fundamental purpose: creating trustworthy, attributable records that support decision-making and regulatory confidence.
The Consumption Jobs: The Hidden Complexity
Electronic signature decisions involve numerous consumption jobs that organizations frequently underestimate:
Evaluation and Selection: “I need to assess when electronic signatures provide genuine value versus when they create unnecessary complexity.”
Implementation and Training: “I need to build electronic signature capability without overwhelming users or compromising data quality.”
Maintenance and Evolution: “I need to keep my signature approach current as regulations evolve and technology advances.”
Integration and Governance: “I need to ensure electronic signatures integrate seamlessly with my broader data integrity strategy.”
These consumption jobs represent the difference between electronic signature systems that users genuinely want to hire and those they grudgingly endure.
The Emotional and Social Dimensions
Electronic signature decisions involve profound emotional and social jobs that traditional compliance approaches ignore:
Confidence: Users want to feel genuinely confident that their signature approach provides appropriate protection, not just regulatory coverage.
Professional Credibility: Quality professionals want signature systems that enhance rather than complicate their ability to ensure data integrity.
Organizational Trust: Executive teams want assurance that their signature approach genuinely protects data integrity rather than creating administrative overhead.
User Acceptance: Operational staff want signature workflows that support rather than impede their work.
The Current Regulatory Landscape: Beyond the Checkbox
Understanding when electronic signatures are required demands a sophisticated appreciation of the regulatory landscape that extends far beyond simple rule application.
FDA 21 CFR Part 11: The Foundation
21 CFR Part 11 establishes that electronic signatures can be equivalent to handwritten signatures when specific conditions are met. However, the regulation’s scope is explicitly limited to situations where signatures are required by predicate rules—the underlying FDA regulations that mandate signatures for specific activities.
The critical insight that most organizations miss: Part 11 doesn’t create new signature requirements. It simply establishes standards for electronic signatures when signatures are already required by other regulations. This distinction is fundamental to proper implementation.
Key Part 11 requirements include:
Unique identification for each individual
Verification of signer identity before assignment
Certification that electronic signatures are legally binding equivalents
Secure signature/record linking to prevent falsification
Comprehensive signature manifestations showing who signed what, when, and why
EU Annex 11: The European Perspective
EU Annex 11 takes a similar approach, requiring that electronic signatures “have the same impact as hand-written signatures”. However, Annex 11 places greater emphasis on risk-based decision making throughout the computerized system lifecycle.
Annex 11’s approach to electronic signatures emphasizes:
GAMP 5 provides the most sophisticated framework for electronic signature decisions, emphasizing risk-based approaches that consider patient safety, product quality, and data integrity throughout the system lifecycle.
GAMP 5’s key principles for electronic signature decisions include:
Risk-based validation approaches
Supplier assessment and leverage
Lifecycle management
Critical thinking application
User requirement specification based on intended use
The Predicate Rule Reality: Where Signatures Are Actually Required
The foundation of any electronic signature decision must be a clear understanding of where signatures are required by predicate rules. These requirements fall into several categories:
Manufacturing Records: Batch records, equipment logbooks, cleaning records where signature accountability is mandated by GMP regulations.
Laboratory Records: Analytical results, method validations, stability studies where analyst and reviewer signatures are required.
Quality Records: Deviation investigations, CAPA records, change controls where signature accountability ensures proper review and approval.
Regulatory Submissions: Clinical data, manufacturing information, safety reports where signatures establish accountability for submitted information.
The critical insight: electronic signatures are only subject to Part 11 requirements when handwritten signatures would be required in the same circumstances.
The Eight-Step Electronic Signature Decision Framework
Applying the Jobs-to-Be-Done universal job map to electronic signature decisions reveals where current approaches systematically fail and how organizations can build genuinely effective signature strategies.
Step 1: Define Context and Purpose
What users need: Clear understanding of the business process, data integrity requirements, regulatory obligations, and decisions the signature will support.
Current reality: Electronic signature decisions often begin with technology evaluation rather than purpose definition, leading to solutions that don’t serve actual needs.
Best practice approach: Begin every electronic signature decision by clearly articulating:
What business process requires authentication
What regulatory requirements mandate signatures
What data integrity risks the signature will address
What decisions the signed record will support
Who will use the signature system and in what context
Step 2: Locate Regulatory Requirements
What users need: Comprehensive understanding of applicable predicate rules, data integrity expectations, and regulatory guidance specific to their process and jurisdiction.
Current reality: Organizations often apply generic interpretations of Part 11 or Annex 11 without understanding the specific predicate rule requirements that drive signature needs.
Best practice approach: Systematically identify:
Specific predicate rules requiring signatures for your process
Applicable data integrity guidance (MHRA, FDA, EMA)
Relevant industry standards (GAMP 5, ICH guidelines)
Jurisdictional requirements for your operations
Industry-specific guidance for your sector
Step 3: Prepare Risk Assessment
What users need: Structured evaluation of risks associated with different signature approaches, considering patient safety, product quality, data integrity, and regulatory compliance.
Current reality: Risk assessments often focus on technical risks rather than the full spectrum of data integrity and business risks associated with signature decisions.
Best practice approach: Develop comprehensive risk assessment considering:
Patient safety implications of signature failure
Product quality risks from inadequate authentication
Data integrity risks from signature system vulnerabilities
Regulatory risks from non-compliant implementation
Business risks from user acceptance and system reliability
Technical risks from system integration and maintenance
Step 4: Confirm Decision Criteria
What users need: Clear criteria for evaluating signature options, with appropriate weighting for different risk factors and user needs.
Current reality: Decision criteria often emphasize technical features over fundamental fitness for purpose, leading to over-engineered or under-protective solutions.
Best practice approach: Establish explicit criteria addressing:
Regulatory compliance requirements
Data integrity protection level needed
User experience and adoption requirements
Technical integration and maintenance needs
Cost-benefit considerations
Long-term sustainability and evolution capability
Step 5: Execute Risk Analysis
What users need: Systematic comparison of signature options against established criteria, with clear rationale for recommendations.
Current reality: Risk analysis often becomes feature comparison rather than genuine assessment of how different approaches serve the jobs users need accomplished.
Best practice approach: Conduct structured analysis that:
Evaluates each option against established criteria
Considers interdependencies with other systems and processes
Assesses implementation complexity and resource requirements
Projects long-term implications and evolution needs
Documents assumptions and limitations
Provides clear recommendation with supporting rationale
Step 6: Monitor Implementation
What users need: Ongoing validation that the chosen signature approach continues to serve its intended purposes and meets evolving requirements.
Current reality: Organizations often treat electronic signature implementation as a one-time decision rather than an ongoing capability requiring continuous monitoring and adjustment.
Best practice approach: Establish monitoring systems that:
Track signature system performance and reliability
Monitor user adoption and satisfaction
Assess continued regulatory compliance
Evaluate data integrity protection effectiveness
Identify emerging risks or opportunities
Measure business value and return on investment
Step 7: Modify Based on Learning
What users need: Responsive adjustment of signature strategies based on monitoring feedback, regulatory changes, and evolving business needs.
Current reality: Electronic signature systems often become static implementations, updated only when forced by system upgrades or regulatory findings.
Best practice approach: Build adaptive capability that:
Incorporates lessons learned from implementation experience
Adapts to changing business needs and user requirements
Leverages technological advances and industry best practices
Maintains documentation of changes and rationale
Step 8: Conclude with Documentation
What users need: Comprehensive documentation that captures the rationale for signature decisions, supports regulatory inspections, and enables knowledge transfer.
Current reality: Documentation often focuses on technical specifications rather than the risk-based rationale that supports the decisions.
Best practice approach: Create documentation that:
Captures the complete decision rationale and supporting analysis
Documents risk assessments and mitigation strategies
Provides clear procedures for ongoing management
Supports regulatory inspection and audit activities
Enables knowledge transfer and training
Facilitates future reviews and updates
The Risk-Based Decision Tool: Moving Beyond Guesswork
The most critical element of any electronic signature strategy is a robust decision tool that enables consistent, risk-based choices. This tool must address the fundamental question: when do electronic signatures provide genuine value over alternative approaches?
The Electronic Signature Decision Matrix
The decision matrix evaluates six critical dimensions:
Regulatory Requirement Level:
High: Predicate rules explicitly require signatures for this activity
Medium: Regulations require documentation/accountability but don’t specify signature method
Low: Good practice suggests signatures but no explicit regulatory requirement
Data Integrity Risk Level:
High: Data directly impacts patient safety, product quality, or regulatory submissions
Medium: Data supports critical quality decisions but has indirect impact
Low: Data supports operational activities with limited quality impact
Process Criticality:
High: Process failure could result in patient harm, product recall, or regulatory action
Medium: Process failure could impact product quality or regulatory compliance
Low: Process failure would have operational impact but limited quality implications
User Environment Factors:
High: Users are technically sophisticated, work in controlled environments, have dedicated time for signature activities
Medium: Users have moderate technical skills, work in mixed environments, have competing priorities
Low: Users have limited technical skills, work in challenging environments, face significant time pressures
System Integration Requirements:
High: Must integrate with validated systems, requires comprehensive audit trails, needs long-term data integrity
Medium: Moderate integration needs, standard audit trail requirements, medium-term data retention
Low: Limited integration needs, basic documentation requirements, short-term data use
Business Value Potential:
High: Electronic signatures could significantly improve efficiency, reduce errors, or enhance compliance
Medium: Moderate improvements in operational effectiveness or compliance capability
Low: Limited operational or compliance benefits from electronic implementation
Decision Logic Framework
Electronic Signature Strongly Recommended (Score: 15-18 points): All high-risk factors align with strong regulatory requirements and favorable implementation conditions. Electronic signatures provide clear value and are essential for compliance.
Electronic Signature Recommended (Score: 12-14 points): Multiple risk factors support electronic signature implementation, with manageable implementation challenges. Benefits outweigh costs and complexity.
Electronic Signature Optional (Score: 9-11 points): Mixed risk factors with both benefits and challenges present. Decision should be based on specific organizational priorities and capabilities.
Alternative Controls Preferred (Score: 6-8 points): Low regulatory requirements combined with implementation challenges suggest alternative controls may be more appropriate.
Electronic Signature Not Recommended (Score: Below 6 points): Risk factors and implementation challenges outweigh potential benefits. Focus on alternative controls and process improvements.
Implementation Guidance by Decision Category
For Strongly Recommended implementations:
Invest in robust, validated electronic signature systems
Implement comprehensive training and competency programs
Establish rigorous monitoring and maintenance procedures
Plan for long-term system evolution and regulatory changes
Plan for future electronic signature capability as conditions change
Maintain documentation of decision rationale for future reference
Practical Implementation Strategies: Building Genuine Capability
Effective electronic signature implementation requires attention to three critical areas: system design, user capability, and governance frameworks.
System Design Considerations
Electronic signature systems must provide robust identity verification that meets both regulatory requirements and practical user needs. This includes:
Authentication and Authorization:
Multi-factor authentication appropriate to risk level
Role-based access controls that reflect actual job responsibilities
Session management that balances security with usability
Integration with existing identity management systems where possible
Signature Manifestation Requirements:
Regulatory requirements for signature manifestation are explicit and non-negotiable. Systems must capture and display:
Printed name of the signer
Date and time of signature execution
Meaning or purpose of the signature (approval, review, authorship, etc.)
Unique identification linking signature to signer
Tamper-evident presentation in both electronic and printed formats
Audit Trail and Data Integrity:
Electronic signature systems must provide comprehensive audit trails that support both routine operations and regulatory inspections. Essential capabilities include:
Immutable recording of all signature-related activities
Integration with broader system audit trail capabilities
Secure storage and long-term preservation of audit information
Searchable and reportable audit trail data
System Integration and Interoperability:
Electronic signatures rarely exist in isolation. Effective implementation requires:
Seamless integration with existing business applications
Consistent user experience across different systems
Data exchange standards that preserve signature integrity
Backup and disaster recovery capabilities
Migration planning for system upgrades and replacements
Training and Competency Development
User Training Programs: Electronic signature success depends critically on user competency. Effective training programs address:
Regulatory requirements and the importance of signature integrity
Proper use of signature systems and security protocols
Recognition and reporting of signature system problems
Understanding of signature meaning and legal implications
Regular refresher training and competency verification
Administrator and Support Training: System administrators require specialized competency in:
Electronic signature system configuration and maintenance
User account and role management
Audit trail monitoring and analysis
Incident response and problem resolution
Regulatory compliance verification and documentation
Management and Oversight Training: Management personnel need understanding of:
Strategic implications of electronic signature decisions
Risk assessment and mitigation approaches
Regulatory compliance monitoring and reporting
Business continuity and disaster recovery planning
Vendor management and assessment requirements
Governance Framework Development
Policy and Procedure Development: Comprehensive governance requires clear policies addressing:
Electronic signature use cases and approval authorities
User qualification and training requirements
System administration and maintenance procedures
Incident response and problem resolution processes
Periodic review and update procedures
Risk Management Integration: Electronic signature governance must integrate with broader quality risk management:
Regular risk assessment updates reflecting system changes
Integration with change control and configuration management
Vendor assessment and ongoing monitoring
Business continuity and disaster recovery testing
Regulatory compliance monitoring and reporting
Performance Monitoring and Continuous Improvement: Effective governance includes ongoing performance management:
Key performance indicators for signature system effectiveness
User satisfaction and adoption monitoring
System reliability and availability tracking
Regulatory compliance verification and trending
Continuous improvement process and implementation
Building Genuine Capability
The ultimate goal of any electronic signature strategy should be building genuine organizational capability rather than simply satisfying regulatory requirements. This requires a fundamental shift in mindset from compliance theater to value creation.
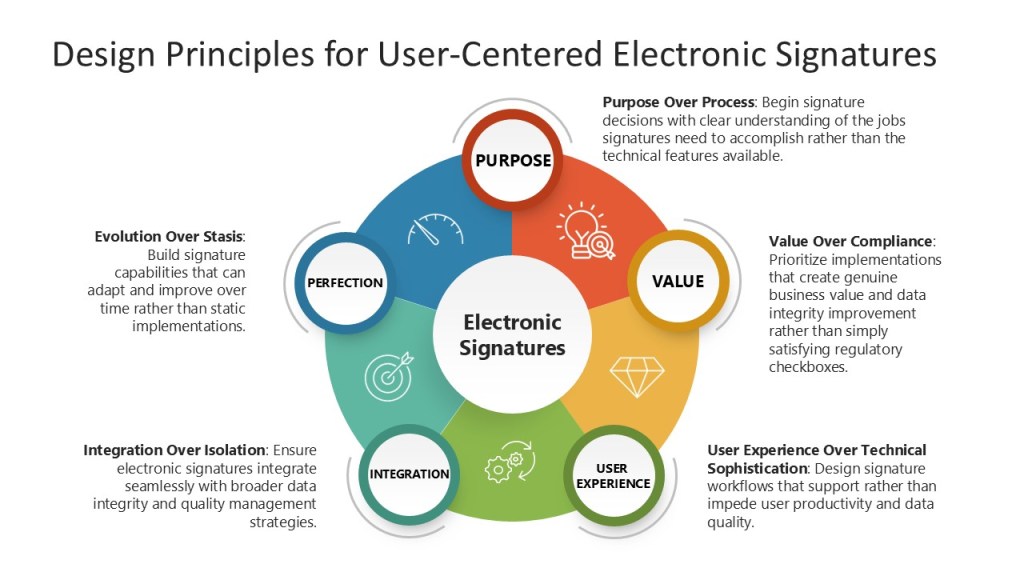
Design Principles for User-Centered Electronic Signatures
Purpose Over Process: Begin signature decisions with clear understanding of the jobs signatures need to accomplish rather than the technical features available.
Value Over Compliance: Prioritize implementations that create genuine business value and data integrity improvement rather than simply satisfying regulatory checkboxes.
User Experience Over Technical Sophistication: Design signature workflows that support rather than impede user productivity and data quality.
Integration Over Isolation: Ensure electronic signatures integrate seamlessly with broader data integrity and quality management strategies.
Evolution Over Stasis: Build signature capabilities that can adapt and improve over time rather than static implementations.
Building Organizational Trust Through Electronic Signatures
Electronic signatures should enhance rather than complicate organizational trust in data integrity. This requires:
Transparency: Users should understand how electronic signatures protect data integrity and support business decisions.
Reliability: Signature systems should work consistently and predictably, supporting rather than impeding daily operations.
Accountability: Electronic signatures should create clear accountability and traceability without overwhelming users with administrative burden.
Competence: Organizations should demonstrate genuine competence in electronic signature implementation and management, not just regulatory compliance.
Future-Proofing Your Electronic Signature Approach
The regulatory and technological landscape for electronic signatures continues to evolve. Organizations need approaches that can adapt to:
Regulatory Evolution: Draft revisions to Annex 11, evolving FDA guidance, and new regulatory requirements in emerging markets.
Technological Advancement: Biometric signatures, blockchain-based authentication, artificial intelligence integration, and mobile signature capabilities.
Business Model Changes: Remote work, cloud-based systems, global operations, and supplier network integration.
User Expectations: Consumerization of technology, mobile-first workflows, and seamless user experiences.
The Path Forward: Hiring Electronic Signatures for Real Jobs
We need to move beyond electronic signature systems that create false confidence while providing no genuine data integrity protection. This happens when organizations optimize for regulatory appearance rather than user needs, creating elaborate signature workflows that nobody genuinely wants to hire.
True electronic signature strategy begins with understanding what jobs users actually need accomplished: establishing reliable accountability, protecting data integrity, enabling efficient workflows, and supporting regulatory confidence. Organizations that design electronic signature approaches around these jobs will develop competitive advantages in an increasingly digital world.
The framework presented here provides a structured approach to making these decisions, but the fundamental insight remains: electronic signatures should not be something organizations implement to satisfy auditors. They should be capabilities that organizations actively seek because they make data integrity demonstrably better.
When we design signature capabilities around the jobs users actually need accomplished—protecting data integrity, enabling accountability, streamlining workflows, and building regulatory confidence—we create systems that enhance rather than complicate our fundamental mission of protecting patients and ensuring product quality.
The choice is clear: continue performing electronic signature compliance theater, or build signature capabilities that organizations genuinely want to hire. In a world where data integrity failures can result in patient harm, product recalls, and regulatory action, only the latter approach offers genuine protection.
Electronic signatures should not be something we implement because regulations require them. They should be capabilities we actively seek because they make us demonstrably better at protecting data integrity and serving patients.
The draft revision of EU GMP Chapter 4 introduces what can only be described as a revolutionary framework for data governance systems. This isn’t merely an update to existing documentation requirements—it is a keystone document that cements the decade long paradigm shift of data governance as the cornerstone of modern pharmaceutical quality systems.
The Genesis of Systematic Data Governance
The most striking aspect of the draft Chapter 4 is the introduction of sections 4.10 through 4.18, which establish data governance systems as mandatory infrastructure within pharmaceutical quality systems. This comprehensive framework emerges from lessons learned during the past decade of data integrity enforcement actions and reflects the reality that modern pharmaceutical manufacturing operates in an increasingly digital environment where traditional documentation approaches are insufficient.
The requirement that regulated users “establish a data governance system integral to the pharmaceutical quality system” moves far beyond the current Chapter 4’s basic documentation requirements. This integration ensures that data governance isn’t treated as an IT afterthought or compliance checkbox, but rather as a fundamental component of how pharmaceutical companies ensure product quality and patient safety. The emphasis on integration with existing pharmaceutical quality systems builds on synergies that I’ve previously discussed in my analysis of how data governance, data quality, and data integrity work together as interconnected pillars.
The requirement for regular documentation and review of data governance arrangements establishes accountability and ensures continuous improvement. This aligns with my observations about risk-based thinking where effective quality systems must anticipate, monitor, respond, and learn from their operational environment.
Comprehensive Data Lifecycle Management
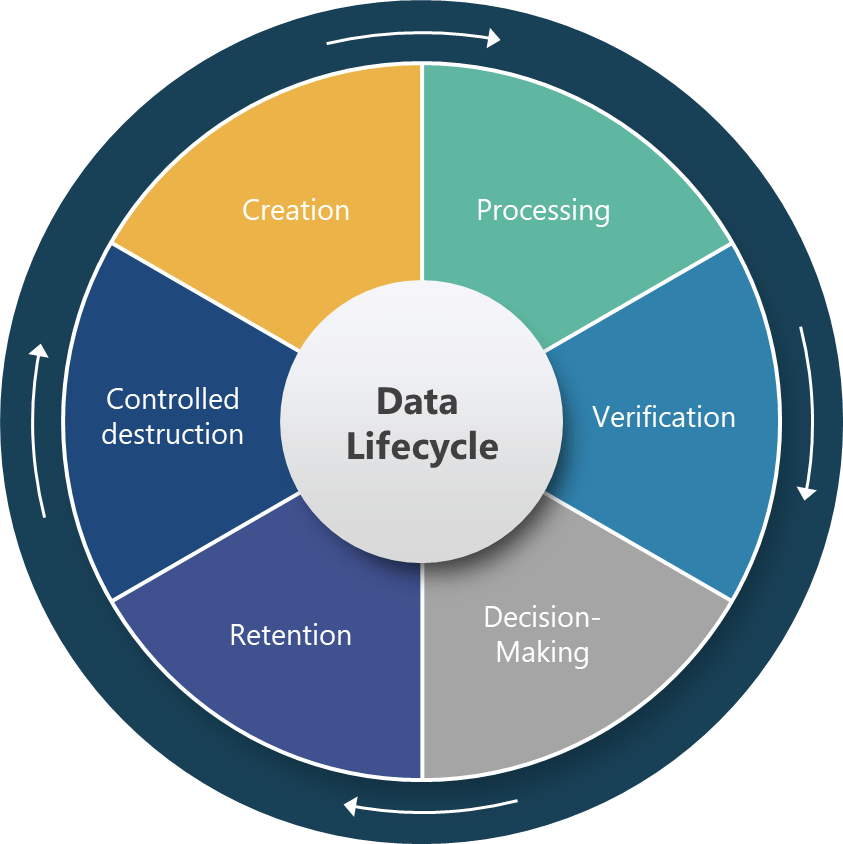
Section 4.12 represents perhaps the most technically sophisticated requirement in the draft, establishing a six-stage data lifecycle framework that covers creation, processing, verification, decision-making, retention, and controlled destruction. This approach acknowledges that data integrity cannot be ensured through point-in-time controls but requires systematic management throughout the entire data journey.
The specific requirement for “reconstruction of all data processing activities” for derived data establishes unprecedented expectations for data traceability and transparency. This requirement will fundamentally change how pharmaceutical companies design their data processing workflows, particularly in areas like process analytical technology (PAT), manufacturing execution systems (MES), and automated batch release systems where raw data undergoes significant transformation before supporting critical quality decisions.
The lifecycle approach also creates direct connections to computerized system validation requirements under Annex 11, as noted in section 4.22. This integration ensures that data governance systems are not separate from, but deeply integrated with, the technical systems that create, process, and store pharmaceutical data. As I’ve discussed in my analysis of computer system validation frameworks, effective validation programs must consider the entire system ecosystem, not just individual software applications.
Risk-Based Data Criticality Assessment
The draft introduces a sophisticated two-dimensional risk assessment framework through section 4.13, requiring organizations to evaluate both data criticality and data risk. Data criticality focuses on the impact to decision-making and product quality, while data risk considers the opportunity for alteration or deletion and the likelihood of detection. This framework provides a scientific basis for prioritizing data protection efforts and designing appropriate controls.
This approach represents a significant evolution from current practices where data integrity controls are often applied uniformly regardless of the actual risk or impact of specific data elements. The risk-based framework allows organizations to focus their most intensive controls on the data that matters most while applying appropriate but proportionate controls to lower-risk information. This aligns with principles I’ve discussed regarding quality risk management under ICH Q9(R1), where structured, science-based approaches reduce subjectivity and improve decision-making.
The requirement to assess “likelihood of detection” introduces a crucial element often missing from traditional data integrity approaches. Organizations must evaluate not only how to prevent data integrity failures but also how quickly and reliably they can detect failures that occur despite preventive controls. This assessment drives requirements for monitoring systems, audit trail analysis capabilities, and incident detection procedures.
Service Provider Oversight and Accountability
Section 4.18 establishes specific requirements for overseeing service providers’ data management policies and risk control strategies. This requirement acknowledges the reality that modern pharmaceutical operations depend heavily on cloud services, SaaS platforms, contract manufacturing organizations, and other external providers whose data management practices directly impact pharmaceutical company compliance.
The risk-based frequency requirement for service provider reviews represents a practical approach that allows organizations to focus oversight efforts where they matter most while ensuring that all service providers receive appropriate attention. For more details on the evolving regulatory expectations around supplier management see the post “draft Annex 11’s supplier oversight requirements“.
The service provider oversight requirement also creates accountability throughout the pharmaceutical supply chain, ensuring that data integrity expectations extend beyond the pharmaceutical company’s direct operations to encompass all entities that handle GMP-relevant data. This approach recognizes that regulatory accountability cannot be transferred to external providers, even when specific activities are outsourced.
Operational Implementation Challenges
The transition to mandatory data governance systems will present significant operational challenges for most pharmaceutical organizations. The requirement for “suitably designed systems, the use of technologies and data security measures, combined with specific expertise” in section 4.14 acknowledges that effective data governance requires both technological infrastructure and human expertise.
Organizations will need to invest in personnel with specialized data governance expertise, implement technology systems capable of supporting comprehensive data lifecycle management, and develop procedures for managing the complex interactions between data governance requirements and existing quality systems. This represents a substantial change management challenge that will require executive commitment and cross-functional collaboration.
The requirement for regular review of risk mitigation effectiveness in section 4.17 establishes data governance as a continuous improvement discipline rather than a one-time implementation project. Organizations must develop capabilities for monitoring the performance of their data governance systems and adjusting controls as risks evolve or new technologies are implemented.
The integration with quality risk management principles throughout sections 4.10-4.22 creates powerful synergies between traditional pharmaceutical quality systems and modern data management practices. This integration ensures that data governance supports rather than competes with existing quality initiatives while providing a systematic framework for managing the increasing complexity of pharmaceutical data environments.
The draft’s emphasis on data ownership throughout the lifecycle in section 4.15 establishes clear accountability that will help organizations avoid the diffusion of responsibility that often undermines data integrity initiatives. Clear ownership models provide the foundation for effective governance, accountability, and continuous improvement.
The concept of “buying down risk” through operational capability development fundamentally depends on addressing the cognitive foundations that underpin effective risk assessment and decision-making. There are three critical systematic vulnerabilities that plague risk management processes: unjustified assumptions, incomplete identification of risks, and inappropriate use of risk assessment tools. These failures represent more than procedural deficiencies—they expose cognitive and knowledge management vulnerabilities that can undermine even the most well-intentioned quality systems.
Unjustified assumptions emerge when organizations rely on historical performance data or familiar process knowledge without adequately considering how changes in conditions, equipment, or supply chains might alter risk profiles. This manifests through anchoring bias, where teams place undue weight on initial information, leading to conclusions like “This process has worked safely for five years, so the risk profile remains unchanged.” Confirmation bias compounds this issue by causing assessors to seek information confirming existing beliefs while ignoring contradictory evidence.
Incomplete risk identification occurs when cognitive limitations and organizational biases inhibit comprehensive hazard recognition. Availability bias leads to overemphasis on dramatic but unlikely events while underestimating more probable but less memorable risks. Additionally, groupthink in risk assessment teams causes initial dissenting voices to be suppressed as consensus builds around preferred conclusions, limiting the scope of risks considered.
Inappropriate use of risk assessment tools represents the third systematic vulnerability, where organizations select methodologies based on familiarity rather than appropriateness for specific decision-making contexts. This includes using overly formal tools for trivial issues, applying generic assessment approaches without considering specific operational contexts, and relying on subjective risk scoring that provides false precision without meaningful insight. The misapplication often leads to risk assessments that fail to add value or clarity because they only superficially address root causes while generating high levels of subjectivity and uncertainty in outputs.
Traditional risk management approaches often focus on methodological sophistication while overlooking the cognitive realities that determine assessment effectiveness. Risk management operates fundamentally as a framework rather than a rigid methodology, providing structural architecture that enables systematic approaches to identifying, assessing, and controlling uncertainties. This framework distinction proves crucial because it recognizes that excellence emerges from the intersection of systematic process design with cognitive support systems that work with, rather than against, human decision-making patterns.
The Minimal Viable Risk Assessment Team: Beyond Compliance Theater
The foundation of cognitive excellence in risk management begins with assembling teams designed for cognitive rigor, knowledge depth, and psychological safety rather than mere compliance box-checking. The minimal viable risk assessment team concept challenges traditional approaches by focusing on four non-negotiable core roles that provide essential cognitive perspectives and knowledge anchors.
The Four Cognitive Anchors
Process Owner: The Reality Anchor represents lived operational experience rather than signature authority. This individual has engaged with the operation within the last 90 days and carries authority to change methods, budgets, and training. Authentic process ownership dismantles assumptions by grounding every risk statement in current operational facts, countering the tendency toward unjustified assumptions that plague many risk assessments.
Molecule Steward: The Patient’s Advocate moves beyond generic subject matter expertise to provide specific knowledge of how the particular product fails and can translate deviations into patient impact. When temperature drifts during freeze-drying, the molecule steward can explain whether a monoclonal antibody will aggregate or merely lose shelf life. Without this anchor, teams inevitably under-score hazards that never appear in generic assessment templates.
Technical System Owner: The Engineering Interpreter bridges the gap between equipment design intentions and operational realities. Equipment obeys physics rather than meeting minutes, and the system owner must articulate functional requirements, design limits, and engineering principles. This role prevents method-focused teams from missing systemic failures where engineering and design flaws could push entire batches outside critical parameters.
Quality Integrator: The Bias Disruptor forces cross-functional dialogue and preserves evidence of decision-making processes. Quality’s mission involves writing assumption logs, challenging confirmation bias, and ensuring dissenting voices are heard. This role maintains knowledge repositories so future teams are not condemned to repeat forgotten errors, directly addressing the knowledge management dimension of systematic risk assessment failure.
Knowledge Accessibility: The Missing Link in Risk Management
The Knowledge Accessibility Index (KAI) provides a systematic framework for evaluating how effectively organizations can access and deploy critical knowledge when decision-making requires specialized expertis. Unlike traditional knowledge management metrics focusing on knowledge creation or storage, the KAI specifically evaluates the availability, retrievability, and usability of knowledge at the point of decision-making.
Four Dimensions of Knowledge Accessibility
Expert Knowledge Availability assesses whether organizations can identify and access subject matter experts when specialized knowledge is required. This includes expert mapping and skill matrices, availability assessment during different operational scenarios, knowledge succession planning, and cross-training coverage for critical capabilities. The pharmaceutical environment demands that a qualified molecule steward be accessible within two hours for critical quality decisions, yet many organizations lack systematic approaches to ensuring this availability.
Knowledge Retrieval Efficiency measures how quickly and effectively teams can locate relevant information when making decisions. This encompasses search functionality effectiveness, knowledge organization and categorization, information architecture alignment with decision-making workflows, and access permissions balancing protection with accessibility. Time to find information represents a critical efficiency indicator that directly impacts the quality of risk assessment outcomes.
Knowledge Quality and Currency evaluates whether accessible knowledge is accurate, complete, and up-to-date through information accuracy verification processes, knowledge update frequency management, source credibility validation mechanisms, and completeness assessment relative to decision-making requirements. Outdated or incomplete knowledge can lead to systematic assessment failures even when expertise appears readily available.
Contextual Applicability assesses whether knowledge can be effectively applied to specific decision-making contexts through knowledge contextualization for operational scenarios, applicability assessment for different situations, integration capabilities with existing processes, and usability evaluation from end-user perspectives. Knowledge that exists but cannot be effectively applied provides little value during critical risk assessment activities.
Effective risk assessment team design fundamentally serves as knowledge preservation, not just compliance fulfillment. Every effective risk team is a living repository of organizational critical process insights, technical know-how, and operational experience. When teams include process owners, technical system engineers, molecule stewards, and quality integrators with deep hands-on familiarity, they collectively safeguard hard-won lessons and tacit knowledge that are often lost during organizational transitions.
Combating organizational forgetting requires intentional, cross-functional team design that fosters active knowledge transfer. When risk teams bring together diverse experts who routinely interact, challenge assumptions, and share context from respective domains, they create dynamic environments where critical information is surfaced, scrutinized, and retained. This living dialogue proves more effective than static records because it allows continuous updating and contextualization of knowledge in response to new challenges, regulatory changes, and operational shifts.
Team design becomes a strategic defense against the silent erosion of expertise that can leave organizations exposed to avoidable risks. By prioritizing teams that embody both breadth and depth of experience, organizations create robust safety nets that catch subtle warning signs, adapt to evolving risks, and ensure critical knowledge endures beyond individual tenure. This transforms collective memory into competitive advantage and foundation for sustained quality.
Cultural Integration: Embedding Cognitive Excellence
The development of truly effective risk management capabilities requires cultural transformation that embeds cognitive excellence principles into organizational DNA. Organizations with strong risk management cultures demonstrate superior capability in preventing quality issues, detecting problems early, and implementing effective corrective actions that address root causes rather than symptoms.
Psychological Safety as Cognitive Infrastructure
Psychological safety creates the foundational environment where personnel feel comfortable challenging assumptions, raising concerns about potential risks, and admitting uncertainty or knowledge limitations. This requires organizational cultures that treat questioning and systematic analysis as valuable contributions rather than obstacles to efficiency. Without psychological safety, the most sophisticated risk assessment methodologies and team compositions cannot overcome the fundamental barrier of information suppression.
Leaders must model vulnerability by sharing personal errors and how systems, not individuals, failed. They must invite dissent early in meetings with questions like “What might we be overlooking?” and reward candor by recognizing people who halt production over questionable trends. Psychological safety converts silent observers into active risk sensors, dramatically improving the effectiveness of knowledge accessibility and risk identification processes.
Structured Decision-Making as Cultural Practice
Excellence in pharmaceutical quality systems requires moving beyond hoping individuals will overcome cognitive limitations through awareness alone. Instead, organizations must design structured decision-making processes that systematically counter known biases while supporting comprehensive risk identification and analysis.
Forced systematic consideration involves checklists, templates, and protocols requiring teams to address specific risk categories and evidence types before reaching conclusions. Rather than relying on free-form discussion influenced by availability bias or groupthink, these tools ensure comprehensive coverage of relevant factors.
Devil’s advocate processes systematically introduce alternative perspectives and challenge preferred conclusions. By assigning specific individuals to argue against prevailing views or identify overlooked risks, organizations counter confirmation bias and overconfidence while identifying blind spots.
Staged decision-making separates risk identification from evaluation, preventing premature closure and ensuring adequate time for comprehensive hazard identification before moving to analysis and control decisions.
Implementation Framework: Building Cognitive Resilience
Phase 1: Knowledge Accessibility Audit
Organizations must begin with systematic knowledge accessibility audits that identify potential vulnerabilities in expertise availability and access. This audit addresses expertise mapping to identify knowledge holders and capabilities, knowledge accessibility assessment evaluating how effectively relevant knowledge can be accessed, knowledge quality evaluation assessing currency and completeness, and cognitive bias vulnerability assessment identifying situations where biases most likely affect conclusions.
For pharmaceutical manufacturing organizations, this audit might assess whether teams can access qualified molecule stewards within two hours for critical quality decisions, whether current system architecture documentation is accessible and comprehensible to risk assessment teams, whether process owners with recent operational experience are available for participation, and whether quality professionals can effectively challenge assumptions and integrate diverse perspectives.
Phase 2: Team Charter and Competence Framework
Moving from compliance theater to protection requires assembling teams with clear charters focused on cognitive rigor rather than checklist completion. An excellent risk team exists to frame, analyze, and communicate uncertainty so businesses can make science-based, patient-centered decisions. Before naming people, organizations must document the decisions teams must enable, the degree of formality those decisions demand, and the resources management will guarantee.
Competence proving rather than role filling ensures each core seat demonstrates documented capabilities. The process owner must have lived the operation recently with authority to change methods and budgets. The molecule steward must understand how specific products fail and translate deviations into patient impact. The technical system owner must articulate functional requirements and design limits. The quality integrator must force cross-functional dialogue and preserve evidence.
Phase 3: Knowledge System Integration
Knowledge-enabled decision making requires structures that make relevant information accessible at decision points while supporting cognitive processes necessary for accurate analysis. This involves structured knowledge capture that explicitly identifies assumptions, limitations, and context rather than simply documenting conclusions. Knowledge validation systems systematically test assumptions embedded in organizational knowledge, including processes for challenging accepted wisdom and updating mental models when new evidence emerges.
Expertise networks connect decision-makers with relevant specialized knowledge when required rather than relying on generalist teams for all assessments. Decision support systems prompt systematic consideration of potential biases and alternative explanations, creating technological infrastructure that supports rather than replaces human cognitive capabilities.
The final phase focuses on embedding cognitive excellence principles into organizational culture through systematic training programs that build both technical competencies and cognitive skills. These programs address not just what tools to use but how to think systematically about complex risk assessment challenges.
Continuous improvement mechanisms systematically analyze risk assessment performance to identify enhancement opportunities and implement improvements in methodologies, training, and support systems. Organizations track prediction accuracy, compare expected versus actual detectability, and feed insights into updated templates and training so subsequent teams start with enhanced capabilities.
Advanced Maturity: Predictive Risk Intelligence
Organizations achieving the highest levels of cognitive excellence implement predictive analytics, real-time bias detection, and adaptive systems that learn from assessment performance. These capabilities enable anticipation of potential risks and bias patterns before they manifest in assessment failures, including systematic monitoring of assessment performance, early warning systems for cognitive failures, and proactive adjustment of assessment approaches based on accumulated experience.
Adaptive learning systems continuously improve organizational capabilities based on performance feedback and changing conditions. These systems identify emerging patterns in risk assessment challenges and automatically adjust methodologies, training programs, and support systems to maintain effectiveness. Organizations at this maturity level contribute to industry knowledge and best practices while serving as benchmarks for other organizations.
From Reactive Compliance to Proactive Capability
The integration of cognitive science insights, knowledge accessibility frameworks, and team design principles creates a transformative approach to pharmaceutical risk management that moves beyond traditional compliance-focused activities toward strategic capability development. Organizations implementing these integrated approaches develop competitive advantages that extend far beyond regulatory compliance.
They build capabilities in systematic decision-making that improve performance across all aspects of pharmaceutical quality management. They create resilient systems that adapt to changing conditions while maintaining consistent effectiveness. Most importantly, they develop cultures of excellence that attract and retain exceptional talent while continuously improving capabilities.
The strategic integration of risk management practices with cultural transformation represents not merely an operational improvement opportunity but a fundamental requirement for sustained success in the evolving pharmaceutical manufacturing environment. Organizations implementing comprehensive risk buy-down strategies through systematic capability development will emerge as industry leaders capable of navigating regulatory complexity while delivering consistent value to patients, stakeholders, and society.
Excellence in this context means designing quality systems that work with human cognitive capabilities rather than against them. This requires integrating knowledge management principles with cognitive science insights to create environments where systematic, evidence-based decision-making becomes natural and sustainable. True elegance in quality system design comes from seamlessly integrating technical excellence with cognitive support, creating systems where the right decisions emerge naturally from the intersection of human expertise and systematic process.
Building Operational Capabilities Through Strategic Risk Management and Cultural Transformation
The Strategic Imperative: Beyond Compliance Theater
The fundamental shift from checklist-driven compliance to sustainable operational excellence grounded in robust risk management culture. Organizations continue to struggle with fundamental capability gaps that manifest as systemic compliance failures, operational disruptions, and ultimately, compromised patient safety.
The Risk Buy-Down Paradigm in Operations
The core challenge here is to build operational capabilities through proactively building systemic competencies that reduce the probability and impact of operational failures over time. Unlike traditional risk mitigation strategies that focus on reactive controls, risk buy-down emphasizes capability development that creates inherent resilience within operational systems.
This paradigm shifts the traditional cost-benefit equation from reactive compliance expenditure to proactive capability investment. Organizations implementing risk buy-down strategies recognize that upfront investments in operational excellence infrastructure generate compounding returns through reduced deviation rates, fewer regulatory observations, improved operational efficiency, and enhanced competitive positioning.
Economic Logic: Investment versus Failure Costs
The financial case for operational capability investment becomes stark when examining failure costs across the pharmaceutical industry. Drug development failures, inclusive of regulatory compliance issues, represent costs ranging from $500 to $900 million per program when accounting for capital costs and failure probabilities. Manufacturing quality failures trigger cascading costs including batch losses, investigation expenses, remediation efforts, regulatory responses, and market disruption.
Pharmaceutical manufacturers continue experiencing fundamental quality system failures despite decades of regulatory enforcement. These failures indicate insufficient investment in underlying operational capabilities, resulting in recurring compliance issues that generate exponentially higher long-term costs than proactive capability development would require.
Organizations successfully implementing risk buy-down strategies demonstrate measurable operational improvements. Companies with strong risk management cultures experience 30% higher likelihood of outperforming competitors while achieving 21% increases in productivity. These performance differentials reflect the compound benefits of systematic capability investment over reactive compliance expenditure.
Just look at the recent whitepaper published by the FDA to see the identified returns to this investment.
Regulatory Intelligence Framework Integration
The regulatory intelligence framework provides crucial foundation for risk buy-down implementation by enabling organizations to anticipate, assess, and proactively address emerging compliance requirements. Rather than responding reactively to regulatory observations, organizations with mature regulatory intelligence capabilities identify systemic capability gaps before they manifest as compliance violations.
Effective regulatory intelligence programs monitor FDA warning letter trends, 483 observations, and enforcement actions to identify patterns indicating capability deficiencies across industry segments. For example, persistent Quality Unit oversight failures across multiple geographic regions indicate fundamental organizational design issues rather than isolated procedural lapses8. This intelligence enables organizations to invest in Quality Unit empowerment, authority structures, and oversight capabilities before experiencing regulatory action.
The integration of regulatory intelligence with risk buy-down strategies creates a proactive capability development cycle where external regulatory trends inform internal capability investments, reducing both regulatory exposure and operational risk while enhancing competitive positioning through superior operational performance.
Culture as the Primary Risk Control
Organizational Culture as Foundational Risk Management
Organizational culture represents the most fundamental risk control mechanism within pharmaceutical operations, directly influencing how quality decisions are made, risks are identified and escalated, and operational excellence is sustained over time. Unlike procedural controls that can be circumvented or technical systems that can fail, culture operates as a pervasive influence that shapes behavior across all organizational levels and operational contexts.
Research demonstrates that organizations with strong risk management cultures are significantly less likely to experience damaging operational risk events and are better positioned to effectively respond when issues do occur.
The foundational nature of culture as a risk control becomes evident when examining quality system failures across pharmaceutical operations. Recent FDA warning letters consistently identify cultural deficiencies underlying technical violations, including insufficient Quality Unit authority, inadequate management commitment to compliance, and systemic failures in risk identification and escalation. These patterns indicate that technical compliance measures alone cannot substitute for robust quality culture.
Quality Culture Impact on Operational Resilience
Quality culture directly influences operational resilience by determining how organizations identify, assess, and respond to quality-related risks throughout manufacturing operations. Organizations with mature quality cultures demonstrate superior capability in preventing quality issues, detecting problems early, and implementing effective corrective actions that address root causes rather than symptoms.
Research in the biopharmaceutical industry reveals that integrating safety and quality cultures creates a unified “Resilience Culture” that significantly enhances organizational ability to sustain high-quality outcomes even under challenging conditions. This resilience culture is characterized by commitment to excellence, customer satisfaction focus, and long-term success orientation that transcends short-term operational pressures.
The operational impact of quality culture manifests through multiple mechanisms. Strong quality cultures promote proactive risk identification where employees at all levels actively surface potential quality concerns before they impact product quality. These cultures support effective escalation processes where quality issues receive appropriate priority regardless of operational pressures. Most importantly, mature quality cultures sustain continuous improvement mindsets where operational challenges become opportunities for systematic capability enhancement.
Dual-Approach Model: Leadership and Employee Ownership
Effective quality culture development requires coordinated implementation of top-down leadership commitment and bottom-up employee ownership, creating organizational alignment around quality principles and operational excellence. This dual-approach model recognizes that sustainable culture transformation cannot be achieved through leadership mandate alone, nor through grassroots initiatives without executive support.
Top-down leadership commitment establishes organizational vision, resource allocation, and accountability structures necessary for quality culture development. Research indicates that leadership commitment is vital for quality culture success and sustainability, with senior management responsible for initiating transformational change, setting quality vision, dedicating resources, communicating progress, and exhibiting visible support. Middle managers and supervisors ensure employees receive direct support and are held accountable to quality values.
Bottom-up employee ownership develops through empowerment, engagement, and competency development that enables staff to integrate quality considerations into daily operations. Organizations achieve employee ownership by incorporating quality into staff orientations, including quality expectations in job descriptions and performance appraisals, providing ongoing training opportunities, granting decision-making authority, and eliminating fear of consequences for quality-related concerns.
The integration of these approaches creates organizational conditions where quality culture becomes self-reinforcing. Leadership demonstrates commitment through resource allocation and decision-making priorities, while employees experience empowerment to make quality-focused decisions without fear of negative consequences for raising concerns or stopping production when quality issues arise.
Culture’s Role in Risk Identification and Response
Mature quality cultures fundamentally alter organizational approaches to risk identification and response by creating psychological safety for surfacing concerns, establishing systematic processes for risk assessment, and maintaining focus on long-term quality outcomes over short-term operational pressures. These cultural characteristics enable organizations to identify and address quality risks before they impact product quality or regulatory compliance.
Risk identification effectiveness depends critically on organizational culture that encourages transparency, values diverse perspectives, and rewards proactive concern identification. Research demonstrates that effective risk cultures promote “speaking up” where employees feel confident raising concerns and leaders demonstrate transparency in decision-making. This cultural foundation enables early risk detection that prevents minor issues from escalating into major quality failures.
Risk response effectiveness reflects cultural values around accountability, continuous improvement, and systematic problem-solving. Organizations with strong risk cultures implement thorough root cause analysis, develop comprehensive corrective and preventive actions, and monitor implementation effectiveness over time. These cultural practices ensure that risk responses address underlying causes rather than symptoms, preventing issue recurrence and building organizational learning capabilities.
The measurement of cultural risk management effectiveness requires systematic assessment of cultural indicators including employee engagement, incident reporting rates, management response to concerns, and the quality of corrective action implementation. Organizations tracking these cultural metrics can identify areas requiring improvement and monitor progress in cultural maturity over time.
Continuous Improvement Culture and Adaptive Capacity
Continuous improvement culture represents a fundamental organizational capability that enables sustained operational excellence through systematic enhancement of processes, systems, and capabilities over time. This culture creates adaptive capacity by embedding improvement mindsets, methodologies, and practices that enable organizations to evolve operational capabilities in response to changing requirements and emerging challenges.
Research demonstrates that continuous improvement culture significantly enhances operational performance through multiple mechanisms. Organizations with strong continuous improvement cultures experience increased employee engagement, higher productivity levels, enhanced innovation, and superior customer satisfaction. These performance improvements reflect the compound benefits of systematic capability development over time.
The development of continuous improvement culture requires systematic investment in employee competencies, improvement methodologies, data collection and analysis capabilities, and organizational learning systems. Organizations achieving mature improvement cultures provide training in improvement methodologies, establish improvement project pipelines, implement measurement systems that track improvement progress, and create recognition systems that reward improvement contributions.
Adaptive capacity emerges from continuous improvement culture through organizational learning mechanisms that capture knowledge from improvement projects, codify successful practices, and disseminate learning across the organization. This learning capability enables organizations to build institutional knowledge that improves response effectiveness to future challenges while preventing recurrence of past issues.
Integration with Regulatory Intelligence and Preventive Action
The integration of continuous improvement methodologies with regulatory intelligence capabilities creates proactive capability development systems that identify and address potential compliance issues before they manifest as regulatory observations. This integration represents advanced maturity in organizational quality management where external regulatory trends inform internal improvement priorities.
Regulatory intelligence provides continuous monitoring of FDA warning letters, 483 observations, enforcement actions, and guidance documents to identify emerging compliance trends and requirements. This intelligence enables organizations to anticipate regulatory expectations and proactively develop capabilities that address potential compliance gaps before they are identified through inspection.
Trending analysis of regulatory observations across industry segments reveals systemic capability gaps that multiple organizations experience. For example, persistent citations for Quality Unit oversight failures indicate industry-wide challenges in Quality Unit empowerment, authority structures, and oversight effectiveness. Organizations with mature regulatory intelligence capabilities use this trending data to assess their own Quality Unit capabilities and implement improvements before experiencing regulatory action.
The implementation of preventive action based on regulatory intelligence creates competitive advantage through superior regulatory preparedness while reducing compliance risk exposure. Organizations systematically analyzing regulatory trends and implementing capability improvements demonstrate regulatory readiness that supports inspection success and enables focus on operational excellence rather than compliance remediation.
The Integration Framework
Aligning Risk Management with Operational Capability Development
The strategic alignment of risk management principles with operational capability development creates synergistic organizational systems where risk identification enhances operational performance while operational excellence reduces risk exposure. This integration requires systematic design of management systems that embed risk considerations into operational processes while using operational data to inform risk management decisions.
Risk-based quality management approaches provide structured frameworks for integrating risk assessment with quality management processes throughout pharmaceutical operations. These approaches move beyond traditional compliance-focused quality management toward proactive systems that identify, assess, and mitigate quality risks before they impact product quality or regulatory compliance.
The implementation of risk-based approaches requires organizational capabilities in risk identification, assessment, prioritization, and mitigation that must be developed through systematic training, process development, and technology implementation. Organizations achieving mature risk-based quality management demonstrate superior performance in preventing quality issues, reducing deviation rates, and maintaining regulatory compliance.
Operational capability development supports risk management effectiveness by creating robust processes, competent personnel, and effective oversight systems that reduce the likelihood of risk occurrence while enhancing response effectiveness when risks do materialize. This capability development includes technical competencies, management systems, and organizational culture elements that collectively create operational resilience.
Efficiency-Excellence-Resilience Nexus
The strategic integration of efficiency, excellence, and resilience objectives creates organizational capabilities that simultaneously optimize resource utilization, maintain high-quality standards, and sustain performance under challenging conditions. This integration challenges traditional assumptions that efficiency and quality represent competing objectives, instead demonstrating that properly designed systems achieve superior performance across all dimensions.
Operational efficiency emerges from systematic elimination of waste, optimization of processes, and effective resource utilization that reduces operational costs while maintaining quality standards.
Operational excellence encompasses consistent achievement of high-quality outcomes through robust processes, competent personnel, and effective management systems.
Operational resilience represents the capability to maintain performance under stress, adapt to changing conditions, and recover effectively from disruptions. Resilience emerges from the integration of efficiency and excellence capabilities with adaptive capacity, redundancy planning, and organizational learning systems that enable sustained performance across varying conditions.
Measurement and Monitoring of Cultural Risk Management
The development of comprehensive measurement systems for cultural risk management enables organizations to track progress, identify improvement opportunities, and demonstrate the business value of culture investments. These measurement systems must capture both quantitative indicators of cultural effectiveness and qualitative assessments of cultural maturity across organizational levels.
Quantitative cultural risk management metrics include employee engagement scores, incident reporting rates, training completion rates, corrective action effectiveness measures, and regulatory compliance indicators. These metrics provide objective measures of cultural performance that can be tracked over time and benchmarked against industry standards.
Qualitative cultural assessment approaches include employee surveys, focus groups, management interviews, and observational assessments that capture cultural nuances not reflected in quantitative metrics. These qualitative approaches provide insights into cultural strengths, improvement opportunities, and the effectiveness of cultural transformation initiatives.
The integration of quantitative and qualitative measurement approaches creates comprehensive cultural assessment capabilities that inform management decision-making while demonstrating progress in cultural maturity. Organizations with mature cultural measurement systems can identify cultural risk indicators early, implement targeted interventions, and track improvement effectiveness over time.
Risk culture measurement frameworks must align with organizational risk appetite, regulatory requirements, and business objectives to ensure relevance and actionability. Effective frameworks establish clear definitions of desired cultural behaviors, implement systematic measurement processes, and create feedback mechanisms that inform continuous improvement in cultural effectiveness.
Common Capability Gaps Revealed Through FDA Observations
Analysis of FDA warning letters and 483 observations reveals persistent capability gaps across pharmaceutical manufacturing operations that reflect systemic weaknesses in organizational design, management systems, and quality culture. These capability gaps manifest as recurring regulatory observations that persist despite repeated enforcement actions, indicating fundamental deficiencies in operational capabilities rather than isolated procedural failures.
Quality Unit oversight failures represent the most frequently cited deficiency in FDA warning letters. These failures encompass insufficient authority to ensure CGMP compliance, inadequate resources for effective oversight, poor documentation practices, and systematic failures in deviation investigation and corrective action implementation. The persistence of Quality Unit deficiencies across multiple geographic regions indicates industry-wide challenges in Quality Unit design and empowerment.
Data integrity violations represent another systematic capability gap revealed through regulatory observations, including falsified records, inappropriate data manipulation, deleted electronic records, and inadequate controls over data generation and review. These violations indicate fundamental weaknesses in data governance systems, personnel training, and organizational culture around data integrity principles.
Deviation investigation and corrective action deficiencies appear consistently across FDA warning letters, reflecting inadequate capabilities in root cause analysis, corrective action development, and implementation effectiveness monitoring. These deficiencies indicate systematic weaknesses in problem-solving methodologies, investigation competencies, and management systems for tracking corrective action effectiveness.
Manufacturing process control deficiencies including inadequate validation, insufficient process monitoring, and poor change control implementation represent persistent capability gaps that directly impact product quality and regulatory compliance. These deficiencies reflect inadequate technical capabilities, insufficient management oversight, and poor integration between manufacturing and quality systems.
GMP Culture Translation to Operational Resilience
The five pillars of GMP – People, Product, Process, Procedures, and Premises – provide comprehensive framework for organizational capability development that addresses all aspects of pharmaceutical manufacturing operations. Effective GMP culture ensures that each pillar receives appropriate attention and investment while maintaining integration across all operational elements.
Personnel competency development represents the foundational element of GMP culture, encompassing technical training, quality awareness, regulatory knowledge, and continuous learning capabilities that enable employees to make appropriate quality decisions across varying operational conditions. Organizations with mature GMP cultures invest systematically in personnel development while creating career advancement opportunities that retain quality expertise.
Process robustness and validation ensure that manufacturing operations consistently produce products meeting quality specifications while providing confidence in process capability under normal operating conditions. GMP culture emphasizes process understanding, validation effectiveness, and continuous monitoring that enables proactive identification and resolution of process issues before they impact product quality.
Documentation systems and data integrity support all aspects of GMP implementation by providing objective evidence of compliance with regulatory requirements while enabling effective investigation and corrective action when issues occur. Mature GMP cultures emphasize documentation accuracy, completeness, and accessibility while implementing controls that prevent data integrity issues.
Risk-Based Quality Management as Operational Capability
Risk-based quality management represents advanced organizational capability that integrates risk assessment principles with quality management processes to create proactive systems that prevent quality issues while optimizing resource allocation. This capability enables organizations to focus quality oversight activities on areas with greatest potential impact while maintaining comprehensive quality assurance across all operations.
The implementation of risk-based quality management requires organizational capabilities in risk identification, assessment, prioritization, and mitigation that must be developed through systematic training, process development, and technology implementation. Organizations achieving mature risk-based capabilities demonstrate superior performance in preventing quality issues, reducing deviation rates, and maintaining regulatory compliance efficiency.
Critical process identification and control strategy development represent core competencies in risk-based quality management that enable organizations to focus resources on processes with greatest potential impact on product quality. These competencies require deep process understanding, risk assessment capabilities, and systematic approaches to control strategy optimization.
Continuous monitoring and trending analysis capabilities enable organizations to identify emerging quality risks before they impact product quality while providing data for systematic improvement of risk management effectiveness. These capabilities require data collection systems, analytical competencies, and management processes that translate monitoring results into proactive risk mitigation actions.
Supplier Management and Third-Party Risk Capabilities
Supplier management and third-party risk management represent critical organizational capabilities that directly impact product quality, regulatory compliance, and operational continuity. The complexity of pharmaceutical supply chains requires sophisticated approaches to supplier qualification, performance monitoring, and risk mitigation that go beyond traditional procurement practices.
Supplier qualification processes must assess not only technical capabilities but also quality culture, regulatory compliance history, and risk management effectiveness of potential suppliers. This assessment requires organizational capabilities in audit planning, execution, and reporting that provide confidence in supplier ability to meet pharmaceutical quality requirements consistently.
Performance monitoring systems must track supplier compliance with quality requirements, delivery performance, and responsiveness to quality issues over time. These systems require data collection capabilities, analytical competencies, and escalation processes that enable proactive management of supplier performance issues before they impact operations.
Risk mitigation strategies must address potential supply disruptions, quality failures, and regulatory compliance issues across the supplier network. Effective risk mitigation requires contingency planning, alternative supplier development, and inventory management strategies that maintain operational continuity while ensuring product quality.
The integration of supplier management with internal quality systems creates comprehensive quality assurance that extends across the entire value chain while maintaining accountability for product quality regardless of manufacturing location or supplier involvement. This integration requires organizational capabilities in supplier oversight, quality agreement management, and cross-functional coordination that ensure consistent quality standards throughout the supply network.
Implementation Roadmap for Cultural Risk Management Development
Staged Approach to Cultural Risk Management Development
The implementation of cultural risk management requires systematic, phased approach that builds organizational capabilities progressively while maintaining operational continuity and regulatory compliance. This staged approach recognizes that cultural transformation requires sustained effort over extended timeframes while providing measurable progress indicators that demonstrate value and maintain organizational commitment.
Phase 1: Foundation Building and Assessment establishes baseline understanding of current culture state, identifies immediate improvement opportunities, and creates infrastructure necessary for systematic cultural development. This phase includes comprehensive cultural assessment, leadership commitment establishment, initial training program development, and quick-win implementation that demonstrates early value from cultural investment.
Cultural assessment activities encompass employee surveys, management interviews, process observations, and regulatory compliance analysis that provide comprehensive understanding of current cultural strengths and improvement opportunities. These assessments establish baseline measurements that enable progress tracking while identifying specific areas requiring focused attention during subsequent phases.
Leadership commitment development ensures that senior management understands cultural transformation requirements, commits necessary resources, and demonstrates visible support for cultural change initiatives. This commitment includes resource allocation, communication of cultural expectations, and integration of cultural objectives into performance management systems.
Phase 2: Capability Development and System Implementation focuses on building specific competencies, implementing systematic processes, and creating organizational infrastructure that supports sustained cultural improvement. This phase includes comprehensive training program rollout, process improvement implementation, measurement system development, and initial culture champion network establishment.
Training program implementation provides employees with knowledge, skills, and tools necessary for effective participation in cultural transformation while creating shared understanding of quality expectations and risk management principles. These programs must be tailored to specific roles and responsibilities while maintaining consistency in core cultural messages.
Process improvement implementation creates systematic approaches to risk identification, assessment, and mitigation that embed cultural values into daily operations. These processes include structured problem-solving methodologies, escalation procedures, and continuous improvement practices that reinforce cultural expectations through routine operational activities.
Phase 3: Integration and Sustainment emphasizes cultural embedding, performance optimization, and continuous improvement capabilities that ensure long-term cultural effectiveness. This phase includes advanced measurement system implementation, culture champion network expansion, and systematic review processes that maintain cultural momentum over time.
Leadership Engagement Strategies for Sustainable Change
Leadership engagement represents the most critical factor in successful cultural transformation, requiring systematic strategies that ensure consistent leadership behavior, effective communication, and sustained commitment throughout the transformation process. Effective leadership engagement creates organizational conditions where cultural change becomes self-reinforcing while providing clear direction and resources necessary for transformation success.
Visible Leadership Commitment requires leaders to demonstrate cultural values through daily decisions, resource allocation priorities, and personal behavior that models expected cultural norms. This visibility includes regular communication of cultural expectations, participation in cultural activities, and recognition of employees who exemplify desired cultural behaviors.
Leadership communication strategies must provide clear, consistent messages about cultural expectations while demonstrating transparency in decision-making and responsiveness to employee concerns. Effective communication includes regular updates on cultural progress, honest discussion of challenges, and celebration of cultural achievements that reinforce the value of cultural investment.
Leadership Development Programs ensure that managers at all levels possess competencies necessary for effective cultural leadership including change management skills, coaching capabilities, and performance management approaches that support cultural transformation. These programs must be ongoing rather than one-time events to ensure sustained leadership effectiveness.
Change management competencies enable leaders to guide employees through cultural transformation while addressing resistance, maintaining morale, and sustaining momentum throughout extended change processes. These competencies include stakeholder engagement, communication planning, and resistance management approaches that facilitate smooth cultural transitions.
Accountability Systems ensure that leaders are held responsible for cultural outcomes within their areas of responsibility while providing support and resources necessary for cultural success. These systems include cultural metrics integration into performance management systems, regular cultural assessment processes, and recognition programs that reward effective cultural leadership.
Training and Development Frameworks
Comprehensive training and development frameworks provide employees with competencies necessary for effective participation in risk-based quality culture while creating organizational learning capabilities that support continuous cultural improvement. These frameworks must be systematic, role-specific, and continuously updated to reflect evolving regulatory requirements and organizational capabilities.
Foundational Training Programs establish basic understanding of quality principles, risk management concepts, and regulatory requirements that apply to all employees regardless of specific role or function. This training creates shared vocabulary and understanding that enables effective cross-functional collaboration while ensuring consistent application of cultural principles.
Quality fundamentals training covers basic concepts including customer focus, process thinking, data-driven decision making, and continuous improvement that form the foundation of quality culture. This training must be interactive, practical, and directly relevant to employee daily responsibilities to ensure engagement and retention.
Risk management training provides employees with capabilities in risk identification, assessment, communication, and escalation that enable proactive risk management throughout operations. This training includes both conceptual understanding and practical tools that employees can apply immediately in their work environment.
Role-Specific Advanced Training develops specialized competencies required for specific positions while maintaining alignment with overall cultural objectives and organizational quality strategy. This training addresses technical competencies, leadership skills, and specialized knowledge required for effective performance in specific roles.
Management training focuses on leadership competencies, change management skills, and performance management approaches that support cultural transformation while achieving operational objectives. This training must be ongoing and include both formal instruction and practical application opportunities.
Technical training ensures that employees possess current knowledge and skills required for effective job performance while maintaining awareness of evolving regulatory requirements and industry best practices. This training includes both initial competency development and ongoing skill maintenance programs.
Continuous Learning Systems create organizational capabilities for identifying training needs, developing training content, and measuring training effectiveness that ensure sustained competency development over time. These systems include needs assessment processes, content development capabilities, and effectiveness measurement approaches that continuously improve training quality.
Metrics and KPIs for Tracking Capability Maturation
Comprehensive measurement systems for cultural capability maturation provide objective evidence of progress while identifying areas requiring additional attention and investment. These measurement systems must balance quantitative indicators with qualitative assessments to capture the full scope of cultural development while providing actionable insights for continuous improvement.
Leading Indicators measure cultural inputs and activities that predict future cultural performance including training completion rates, employee engagement scores, participation in improvement activities, and leadership behavior assessments. These indicators provide early warning of cultural issues while demonstrating progress in cultural development activities.
Employee engagement measurements capture employee commitment to organizational objectives, satisfaction with work environment, and confidence in organizational leadership that directly influence cultural effectiveness. These measurements include regular survey processes, focus group discussions, and exit interview analysis that provide insights into employee perspectives on cultural development.
Training effectiveness indicators track not only completion rates but also competency development, knowledge retention, and application of training content in daily work activities. These indicators ensure that training investments translate into improved job performance and cultural behavior.
Lagging Indicators measure cultural outcomes including quality performance, regulatory compliance, operational efficiency, and customer satisfaction that reflect the ultimate impact of cultural investments. These indicators provide validation of cultural effectiveness while identifying areas where cultural development has not yet achieved desired outcomes.
Quality performance metrics include deviation rates, customer complaints, product recalls, and regulatory observations that directly reflect the effectiveness of quality culture in preventing quality issues. These metrics must be trended over time to identify improvement patterns and areas requiring additional attention.
Operational efficiency indicators encompass productivity measures, cost performance, delivery performance, and resource utilization that demonstrate the operational impact of cultural improvements. These indicators help demonstrate the business value of cultural investments while identifying opportunities for further improvement.
Integrated Measurement Systems combine leading and lagging indicators into comprehensive dashboards that provide management with complete visibility into cultural development progress while enabling data-driven decision making about cultural investments. These systems include automated data collection, trend analysis capabilities, and exception reporting that focus management attention on areas requiring intervention.
Benchmarking capabilities enable organizations to compare their cultural performance against industry standards and best practices while identifying opportunities for improvement. These capabilities require access to industry data, analytical competencies, and systematic comparison processes that inform cultural development strategies.
Future-Facing Implications for the Evolving Regulatory Landscape
Emerging Regulatory Trends and Capability Requirements
The regulatory landscape continues evolving toward increased emphasis on risk-based approaches, data integrity requirements, and organizational culture assessment that require corresponding evolution in organizational capabilities and management approaches. Organizations must anticipate these regulatory developments and proactively develop capabilities that address future requirements rather than merely responding to current regulations.
Enhanced Quality Culture Focus in regulatory inspections requires organizations to demonstrate not only technical compliance but also cultural effectiveness in sustaining quality performance over time. This trend requires development of cultural measurement capabilities, cultural audit processes, and systematic approaches to cultural development that provide evidence of cultural maturity to regulatory inspectors.
Risk-based inspection approaches focus regulatory attention on areas with greatest potential risk while requiring organizations to demonstrate effective risk management capabilities throughout their operations. This evolution requires mature risk assessment capabilities, comprehensive risk mitigation strategies, and systematic documentation of risk management effectiveness.
Technology Integration and Cultural Adaptation
Technology integration in pharmaceutical manufacturing creates new opportunities for operational excellence while requiring cultural adaptation that maintains human oversight and decision-making capabilities in increasingly automated environments. Organizations must develop cultural approaches that leverage technology capabilities while preserving the human judgment and oversight essential for quality decision-making.
Digital quality systems enable real-time monitoring, advanced analytics, and automated decision support that enhance quality management effectiveness while requiring new competencies in system operation, data interpretation, and technology-assisted decision making. Cultural adaptation must ensure that technology enhances rather than replaces human quality oversight capabilities.
Data Integrity in Digital Environments requires sophisticated understanding of electronic systems, data governance principles, and cybersecurity requirements that go beyond traditional paper-based quality systems. Cultural development must emphasize data integrity principles that apply across both electronic and paper systems while building competencies in digital data management.
Building Adaptive Organizational Capabilities
The increasing pace of change in regulatory requirements, technology capabilities, and market conditions requires organizational capabilities that enable rapid adaptation while maintaining operational stability and quality performance. These adaptive capabilities must be embedded in organizational culture and management systems to ensure sustained effectiveness across changing conditions.
Learning Organization Capabilities enable systematic capture, analysis, and dissemination of knowledge from operational experience, regulatory changes, and industry developments that inform continuous organizational improvement. These capabilities include knowledge management systems, learning processes, and cultural practices that promote organizational learning and adaptation.
Scenario planning and contingency management capabilities enable organizations to anticipate potential future conditions and develop response strategies that maintain operational effectiveness across varying circumstances. These capabilities require analytical competencies, strategic planning processes, and risk management approaches that address uncertainty systematically.
Change Management Excellence encompasses systematic approaches to organizational change that minimize disruption while maximizing adoption of new capabilities and practices. These capabilities include change planning, stakeholder engagement, communication strategies, and performance management approaches that facilitate smooth organizational transitions.
Resilience building requires organizational capabilities that enable sustained performance under stress, rapid recovery from disruptions, and systematic strengthening of organizational capabilities based on experience with challenges. These capabilities encompass redundancy planning, crisis management, business continuity, and systematic approaches to capability enhancement based on lessons learned.
The future pharmaceutical manufacturing environment will require organizations that combine operational excellence with adaptive capability, regulatory intelligence with proactive compliance, and technical competence with robust quality culture. Organizations successfully developing these integrated capabilities will achieve sustainable competitive advantage while contributing to improved patient outcomes through reliable access to high-quality pharmaceutical products.
The strategic integration of risk management practices with cultural transformation represents not merely an operational improvement opportunity but a fundamental requirement for sustained success in the evolving pharmaceutical manufacturing environment. Organizations implementing comprehensive risk buy-down strategies through systematic capability development will emerge as industry leaders capable of navigating regulatory complexity while delivering consistent value to patients, stakeholders, and society.
The current state of periodic reviews in most pharmaceutical organizations is, to put it charitably, underwhelming. Annual checkbox exercises where teams dutifully document that “the system continues to operate as intended” while avoiding any meaningful analysis of actual system performance, emerging risks, or validation gaps. I’ve seen periodic reviews that consist of little more than confirming the system is still running and updating a few SOPs. This approach might have survived regulatory scrutiny in simpler times, but Section 14 of the draft Annex 11 obliterates this compliance theater and replaces it with rigorous, systematic, and genuinely valuable system intelligence.
The new requirements in the draft Annex 11 Section 14: Periodic Review don’t just raise the bar—they relocate it to a different universe entirely. Where the 2011 version suggested that systems “should be periodically evaluated,” the draft mandates comprehensive, structured, and consequential reviews that must demonstrate continued fitness for purpose and validated state. Organizations that have treated periodic reviews as administrative burdens are about to discover they’re actually the foundation of sustainable digital compliance.
The Philosophical Revolution: From Static Assessment to Dynamic Intelligence
The fundamental transformation in Section 14 reflects a shift from viewing computerized systems as static assets that require occasional maintenance to understanding them as dynamic, evolving components of complex pharmaceutical operations that require continuous intelligence and adaptive management. This philosophical change acknowledges several uncomfortable realities that the industry has long ignored.
First, modern computerized systems never truly remain static. Cloud platforms undergo continuous updates. SaaS providers deploy new features regularly. Integration points evolve. User behaviors change. Regulatory requirements shift. Security threats emerge. Business processes adapt. The fiction that a system can be validated once and then monitored through cursory annual reviews has become untenable in environments where change is the only constant.
Second, the interconnected nature of modern pharmaceutical operations means that changes in one system ripple through entire operational ecosystems in ways that traditional periodic reviews rarely capture. A seemingly minor update to a laboratory information management system might affect data flows to quality management systems, which in turn impact batch release processes, which ultimately influence regulatory reporting. Section 14 acknowledges this complexity by requiring assessment of combined effects across multiple systems and changes.
Third, the rise of data integrity as a central regulatory concern means that periodic reviews must evolve beyond functional assessment to include sophisticated analysis of data handling, protection, and preservation throughout increasingly complex digital environments. This requires capabilities that most current periodic review processes simply don’t possess.
Section 14.1 establishes the foundational requirement that “computerised systems should be subject to periodic review to verify that they remain fit for intended use and in a validated state.” This language moves beyond the permissive “should be evaluated” of the current regulation to establish periodic review as a mandatory demonstration of continued compliance rather than optional best practice.
The requirement that reviews verify systems remain “fit for intended use” introduces a performance-based standard that goes beyond technical functionality to encompass business effectiveness, regulatory adequacy, and operational sustainability. Systems might continue to function technically while becoming inadequate for their intended purposes due to changing regulatory requirements, evolving business processes, or emerging security threats.
Similarly, the requirement to verify systems remain “in a validated state” acknowledges that validation is not a permanent condition but a dynamic state that can be compromised by changes, incidents, or evolving understanding of system risks and requirements. This creates an ongoing burden of proof that validation status is actively maintained rather than passively assumed.
The Twelve Pillars of Comprehensive System Intelligence
Section 14.2 represents perhaps the most significant transformation in the entire draft regulation by establishing twelve specific areas that must be addressed in every periodic review. This prescriptive approach eliminates the ambiguity that has allowed organizations to conduct superficial reviews while claiming regulatory compliance.
The requirement to assess “changes to hardware and software since the last review” acknowledges that modern systems undergo continuous modification through patches, updates, configuration changes, and infrastructure modifications. Organizations must maintain comprehensive change logs and assess the cumulative impact of all modifications on system validation status, not just changes that trigger formal change control processes.
“Changes to documentation since the last review” recognizes that documentation drift—where procedures, specifications, and validation documents become disconnected from actual system operation—represents a significant compliance risk. Reviews must identify and remediate documentation gaps that could compromise operational consistency or regulatory defensibility.
The requirement to evaluate “combined effect of multiple changes” addresses one of the most significant blind spots in traditional change management approaches. Individual changes might be assessed and approved through formal change control processes, but their collective impact on system performance, validation status, and operational risk often goes unanalyzed. Section 14 requires systematic assessment of how multiple changes interact and whether their combined effect necessitates revalidation activities.
“Undocumented or not properly controlled changes” targets one of the most persistent compliance failures in pharmaceutical operations. Despite robust change control procedures, systems inevitably undergo modifications that bypass formal processes. These might include emergency fixes, vendor-initiated updates, configuration drift, or unauthorized user modifications. Periodic reviews must actively hunt for these changes and assess their impact on validation status.
The focus on “follow-up on CAPAs” integrates corrective and preventive actions into systematic review processes, ensuring that identified issues receive appropriate attention and that corrective measures prove effective over time. This creates accountability for CAPA effectiveness that extends beyond initial implementation to long-term performance.
Requirements to assess “security incidents and other incidents” acknowledge that system security and reliability directly impact validation status and regulatory compliance. Organizations must evaluate whether incidents indicate systematic vulnerabilities that require design changes, process improvements, or enhanced controls.
“Non-conformities” assessment requires systematic analysis of deviations, exceptions, and other performance failures to identify patterns that might indicate underlying system inadequacies or operational deficiencies requiring corrective action.
The mandate to review “applicable regulatory updates” ensures that systems remain compliant with evolving regulatory requirements rather than becoming progressively non-compliant as guidance documents are revised, new regulations are promulgated, or inspection practices evolve.
“Audit trail reviews and access reviews” elevates these critical data integrity activities from routine operational tasks to strategic compliance assessments that must be evaluated for effectiveness, completeness, and adequacy as part of systematic periodic review.
Requirements for “supporting processes” assessment acknowledge that computerized systems operate within broader procedural and organizational contexts that directly impact their effectiveness and compliance. Changes to training programs, quality systems, or operational procedures might affect system validation status even when the systems themselves remain unchanged.
The focus on “service providers and subcontractors” reflects the reality that modern pharmaceutical operations depend heavily on external providers whose performance directly impacts system compliance and effectiveness. As I discussed in my analysis of supplier management requirements, organizations cannot outsource accountability for system compliance even when they outsource system operation.
Finally, the requirement to assess “outsourced activities” ensures that organizations maintain oversight of all system-related functions regardless of where they are performed or by whom, acknowledging that regulatory accountability cannot be transferred to external providers.
Review Area
Primary Objective
Key Focus Areas
Hardware/Software Changes
Track and assess all system modifications
Change logs, patch management, infrastructure updates, version control
Documentation Changes
Ensure documentation accuracy and currency
Document version control, procedure updates, specification accuracy, training materials
Combined Change Effects
Evaluate cumulative change impact
Cumulative change impact, system interactions, validation status implications
Undocumented Changes
Identify and control unmanaged changes
Change detection, impact assessment, process gap identification, control improvements
CAPA Follow-up
Verify corrective action effectiveness
CAPA effectiveness, root cause resolution, preventive measure adequacy, trend analysis
Section 14.3 establishes a risk-based approach to periodic review frequency that moves beyond arbitrary annual schedules to systematic assessment of when reviews are needed based on “the system’s potential impact on product quality, patient safety and data integrity.” This approach aligns with broader pharmaceutical industry trends toward risk-based regulatory strategies while acknowledging that different systems require different levels of ongoing attention.
The risk-based approach requires organizations to develop sophisticated risk assessment capabilities that can evaluate system criticality across multiple dimensions simultaneously. A laboratory information management system might have high impact on product quality and data integrity but lower direct impact on patient safety, suggesting different review priorities and frequencies compared to a clinical trial management system or manufacturing execution system.
Organizations must document their risk-based frequency decisions and be prepared to defend them during regulatory inspections. This creates pressure for systematic, scientifically defensible risk assessment methodologies rather than intuitive or political decision-making about resource allocation.
The risk-based approach also requires dynamic adjustment as system characteristics, operational contexts, or regulatory environments change. A system that initially warranted annual reviews might require more frequent attention if it experiences reliability problems, undergoes significant changes, or becomes subject to enhanced regulatory scrutiny.
FREQUENCY: Semi-annually DEPTH: Focused+ (critical areas with simplified analysis) RESOURCES: Quality lead + SME support EXAMPLES: Critical Parameter Monitoring, Sterility Testing Systems, Release Testing Platforms FOCUS: Performance validation, data integrity verification, regulatory compliance
Medium Criticality Systems
High Complexity
Medium Complexity
Low Complexity
FREQUENCY: Semi-annually DEPTH: Standard (structured assessment) RESOURCES: Cross-functional team EXAMPLES: Enterprise Resource Planning, Advanced Analytics Platforms, Multi-system Integrations FOCUS: System integration assessment, change impact analysis, performance optimization
FREQUENCY: Annually DEPTH: Standard (balanced assessment) RESOURCES: Small team EXAMPLES: Training Management Systems, Calibration Management, Standard Laboratory Instruments FOCUS: Operational effectiveness, compliance maintenance, trend monitoring
FREQUENCY: Annually DEPTH: Focused (key areas only) RESOURCES: Individual reviewer + occasional SME EXAMPLES: Simple Data Loggers, Basic Trending Tools, Standard Office Applications FOCUS: Basic functionality verification, minimal compliance checking
Documentation and Analysis: From Checklists to Intelligence Reports
Section 14.4 transforms documentation requirements from simple record-keeping to sophisticated analytical reporting that must “document the review, analyze the findings and identify consequences, and be implemented to prevent any reoccurrence.” This language establishes periodic reviews as analytical exercises that generate actionable intelligence rather than administrative exercises that produce compliance artifacts.
The requirement to “analyze the findings” means that reviews must move beyond simple observation to systematic evaluation of what findings mean for system performance, validation status, and operational risk. This analysis must be documented in ways that demonstrate analytical rigor and support decision-making about system improvements, validation activities, or operational changes.
“Identify consequences” requires forward-looking assessment of how identified issues might affect future system performance, compliance status, or operational effectiveness. This prospective analysis helps organizations prioritize corrective actions and allocate resources effectively while demonstrating proactive risk management.
The mandate to implement measures “to prevent any reoccurrence” establishes accountability for corrective action effectiveness that extends beyond traditional CAPA processes to encompass systematic prevention of issue recurrence through design changes, process improvements, or enhanced controls.
These documentation requirements create significant implications for periodic review team composition, analytical capabilities, and reporting systems. Organizations need teams with sufficient technical and regulatory expertise to conduct meaningful analysis and systems capable of supporting sophisticated analytical reporting.
Integration with Quality Management Systems: The Nervous System Approach
Perhaps the most transformative aspect of Section 14 is its integration with broader quality management system activities. Rather than treating periodic reviews as isolated compliance exercises, the new requirements position them as central intelligence-gathering activities that inform broader organizational decision-making about system management, validation strategies, and operational improvements.
This integration means that periodic review findings must flow systematically into change control processes, CAPA systems, validation planning, supplier management activities, and regulatory reporting. Organizations can no longer conduct periodic reviews in isolation from other quality management activities—they must demonstrate that review findings drive appropriate organizational responses across all relevant functional areas.
The integration also means that periodic review schedules must align with other quality management activities including management reviews, internal audits, supplier assessments, and regulatory inspections. Organizations need coordinated calendars that ensure periodic review findings are available to inform these other activities while avoiding duplicative or conflicting assessment activities.
Technology Requirements: Beyond Spreadsheets and SharePoint
The analytical and documentation requirements of Section 14 push most current periodic review approaches beyond their technological limits. Organizations relying on spreadsheets, email coordination, and SharePoint collaboration will find these tools inadequate for systematic multi-system analysis, trend identification, and integrated reporting required by the new regulation.
Effective implementation requires investment in systems capable of aggregating data from multiple sources, supporting collaborative analysis, maintaining traceability throughout review processes, and generating reports suitable for regulatory presentation. These might include dedicated GRC (Governance, Risk, and Compliance) platforms, advanced quality management systems, or integrated validation lifecycle management tools.
The technology requirements extend to underlying system monitoring and data collection capabilities. Organizations need systems that can automatically collect performance data, track changes, monitor security events, and maintain audit trails suitable for periodic review analysis. Manual data collection approaches become impractical when reviews must assess twelve specific areas across multiple systems on risk-based schedules.
Resource and Competency Implications: Building Analytical Capabilities
Section 14’s requirements create significant implications for organizational capabilities and resource allocation. Traditional periodic review approaches that rely on part-time involvement from operational personnel become inadequate for systematic multi-system analysis requiring technical, regulatory, and analytical expertise.
Organizations need dedicated periodic review capabilities that might include full-time coordinators, subject matter expert networks, analytical tool specialists, and management reporting coordinators. These teams need training in analytical methodologies, regulatory requirements, technical system assessment, and organizational change management.
The competency requirements extend beyond technical skills to include systems thinking capabilities that can assess interactions between systems, processes, and organizational functions. Team members need understanding of how changes in one area might affect other areas and how to design analytical approaches that capture these complex relationships.
Comparison with Current Practices: The Gap Analysis
The transformation from current periodic review practices to Section 14 requirements represents one of the largest compliance gaps in the entire draft Annex 11. Most organizations conduct periodic reviews that bear little resemblance to the comprehensive analytical exercises envisioned by the new regulation.
Current practices typically focus on confirming that systems continue to operate and that documentation remains current. Section 14 requires systematic analysis of system performance, validation status, risk evolution, and operational effectiveness across twelve specific areas with documented analytical findings and corrective action implementation.
Current practices often treat periodic reviews as isolated compliance exercises with minimal integration into broader quality management activities. Section 14 requires tight integration with change management, CAPA processes, supplier management, and regulatory reporting.
Current practices frequently rely on annual schedules regardless of system characteristics or operational context. Section 14 requires risk-based frequency determination with documented justification and dynamic adjustment based on changing circumstances.
Current practices typically produce simple summary reports with minimal analytical content. Section 14 requires sophisticated analytical reporting that identifies trends, assesses consequences, and drives organizational decision-making.
GAMP 5 Alignment and Evolution
GAMP 5’s approach to periodic review provides a foundation for implementing Section 14 requirements but requires significant enhancement to meet the new regulatory standards. GAMP 5 recommends periodic review as best practice for maintaining validation throughout system lifecycles and provides guidance on risk-based approaches to frequency determination and scope definition.
However, GAMP 5’s recommendations lack the prescriptive detail and mandatory requirements of Section 14. While GAMP 5 suggests comprehensive system review including technical, procedural, and performance aspects, it doesn’t mandate the twelve specific areas required by Section 14. GAMP 5 recommends formal documentation and analytical reporting but doesn’t establish the specific analytical and consequence identification requirements of the new regulation.
The GAMP 5 emphasis on integration with overall quality management systems aligns well with Section 14 requirements, but organizations implementing GAMP 5 guidance will need to enhance their approaches to meet the more stringent requirements of the draft regulation.
Organizations that have successfully implemented GAMP 5 periodic review recommendations will have significant advantages in transitioning to Section 14 compliance, but they should not assume their current approaches are adequate without careful gap analysis and enhancement planning.
Implementation Strategy: From Current State to Section 14 Compliance
Organizations planning Section 14 implementation must begin with comprehensive assessment of current periodic review practices against the new requirements. This gap analysis should address all twelve mandatory review areas, analytical capabilities, documentation standards, integration requirements, and resource needs.
The implementation strategy should prioritize development of analytical capabilities and supporting technology infrastructure. Organizations need systems capable of collecting, analyzing, and reporting the complex multi-system data required for Section 14 compliance. This typically requires investment in new technology platforms and development of new analytical competencies.
Change management becomes critical for successful implementation because Section 14 requirements represent fundamental changes in how organizations approach system oversight. Stakeholders accustomed to routine annual reviews must be prepared for analytical exercises that might identify significant system issues requiring substantial corrective actions.
Training and competency development programs must address the enhanced analytical and technical requirements of Section 14 while ensuring that review teams understand their integration responsibilities within broader quality management systems.
Organizations should plan phased implementation approaches that begin with pilot programs on selected systems before expanding to full organizational implementation. This allows refinement of procedures, technology, and competencies before deploying across entire system portfolios.
The Final Review Requirement: Planning for System Retirement
Section 14.5 introduces a completely new concept: “A final review should be performed when a computerised system is taken out of use.” This requirement acknowledges that system retirement represents a critical compliance activity that requires systematic assessment and documentation.
The final review requirement addresses several compliance risks that traditional system retirement approaches often ignore. Organizations must ensure that all data preservation requirements are met, that dependent systems continue to operate appropriately, that security risks are properly addressed, and that regulatory reporting obligations are fulfilled.
Final reviews must assess the impact of system retirement on overall operational capabilities and validation status of remaining systems. This requires understanding of system interdependencies that many organizations lack and systematic assessment of how retirement might affect continuing operations.
The final review requirement also creates documentation obligations that extend system compliance responsibilities through the retirement process. Organizations must maintain evidence that system retirement was properly planned, executed, and documented according to regulatory requirements.
Regulatory Implications and Inspection Readiness
Section 14 requirements fundamentally change regulatory inspection dynamics by establishing periodic reviews as primary evidence of continued system compliance and organizational commitment to maintaining validation throughout system lifecycles. Inspectors will expect to see comprehensive analytical reports with documented findings, systematic corrective actions, and clear integration with broader quality management activities.
The twelve mandatory review areas provide inspectors with specific criteria for evaluating periodic review adequacy. Organizations that cannot demonstrate systematic assessment of all required areas will face immediate compliance challenges regardless of overall system performance.
The analytical and documentation requirements create expectations for sophisticated compliance artifacts that demonstrate organizational competency in system oversight and continuous improvement. Superficial reviews with minimal analytical content will be viewed as inadequate regardless of compliance with technical system requirements.
The integration requirements mean that inspectors will evaluate periodic reviews within the context of broader quality management system effectiveness. Disconnected or isolated periodic reviews will be viewed as evidence of inadequate quality system integration and organizational commitment to continuous improvement.
Strategic Implications: Periodic Review as Competitive Advantage
Organizations that successfully implement Section 14 requirements will gain significant competitive advantages through enhanced system intelligence, proactive risk management, and superior operational effectiveness. Comprehensive periodic reviews provide organizational insights that enable better system selection, more effective resource allocation, and proactive identification of improvement opportunities.
The analytical capabilities required for Section 14 compliance support broader organizational decision-making about technology investments, process improvements, and operational strategies. Organizations that develop these capabilities for periodic review purposes can leverage them for strategic planning, performance management, and continuous improvement initiatives.
The integration requirements create opportunities for enhanced organizational learning and knowledge management. Systematic analysis of system performance, validation status, and operational effectiveness generates insights that can improve future system selection, implementation, and management decisions.
Organizations that excel at Section 14 implementation will build reputations for regulatory sophistication and operational excellence that provide advantages in regulatory relationships, business partnerships, and talent acquisition.
The Future of Pharmaceutical System Intelligence
Section 14 represents the evolution of pharmaceutical compliance toward sophisticated organizational intelligence systems that provide real-time insight into system performance, validation status, and operational effectiveness. This evolution acknowledges that modern pharmaceutical operations require continuous monitoring and adaptive management rather than periodic assessment and reactive correction.
The transformation from compliance theater to genuine system intelligence creates opportunities for pharmaceutical organizations to leverage their compliance investments for strategic advantage while ensuring robust regulatory compliance. Organizations that embrace this transformation will build sustainable competitive advantages through superior system management and operational effectiveness.
However, the transformation also creates significant implementation challenges that will test organizational commitment to compliance excellence. Organizations that attempt to meet Section 14 requirements through incremental enhancement of current practices will likely fail to achieve adequate compliance or realize strategic benefits.
Success requires fundamental reimagining of periodic review as organizational intelligence activity that provides strategic value while ensuring regulatory compliance. This requires investment in technology, competencies, and processes that extend well beyond traditional compliance requirements but provide returns through enhanced operational effectiveness and strategic insight.
Summary Comparison: The New Landscape of Periodic Review
Aspect
Draft Annex 11 Section 14 (2025)
Current Annex 11 (2011)
GAMP 5 Recommendations
Regulatory Mandate
Mandatory periodic reviews to verify system remains “fit for intended use” and “in validated state”
Systems “should be periodically evaluated” – less prescriptive mandate
Strongly recommended as best practice for maintaining validation throughout lifecycle
Scope of Review
12 specific areas mandated including changes, supporting processes, regulatory updates, security incidents
General areas listed: functionality, deviation records, incidents, problems, upgrade history, performance, reliability, security
Comprehensive system review including technical, procedural, and performance aspects
Risk-Based Approach
Frequency based on risk assessment of system impact on product quality, patient safety, data integrity
Risk-based approach implied but not explicitly required
Core principle – review depth and frequency based on system criticality and risk
Documentation Requirements
Reviews must be documented, findings analyzed, consequences identified, prevention measures implemented
Implicit documentation requirement but not explicitly detailed
Formal documentation recommended with structured reporting
Integration with Quality System
Integrated with audits, inspections, CAPA, incident management, security assessments
Limited integration requirements specified
Integrated with overall quality management system and change control
Follow-up Actions
Findings must be analyzed to identify consequences and prevent recurrence
No specific follow-up action requirements
Action plans for identified issues with tracking to closure
Final System Review
Final review mandated when system taken out of use
No final review requirement specified
Retirement planning and data preservation activities
The transformation represented by Section 14 marks the end of periodic review as administrative burden and its emergence as strategic organizational capability. Organizations that recognize and embrace this transformation will build sustainable competitive advantages while ensuring robust regulatory compliance. Those that resist will find themselves increasingly disadvantaged in regulatory relationships and operational effectiveness as the pharmaceutical industry evolves toward more sophisticated digital compliance approaches.
Annex 11 Section 14 Integration: Computerized System Intelligence as the Foundation of CPV Excellence
The sophisticated framework for Continuous Process Verification (CPV) methodology and tool selection outlined in this post intersects directly with the revolutionary requirements of Draft Annex 11 Section 14 on periodic review. While CPV focuses on maintaining process validation through statistical monitoring and adaptive control, Section 14 ensures that the computerized systems underlying CPV programs remain in validated states and continue to generate trustworthy data throughout their operational lifecycles.
This intersection represents a critical compliance nexus where process validation meets system validation, creating dependencies that pharmaceutical organizations must understand and manage systematically. The failure to maintain computerized systems in validated states directly undermines CPV program integrity, while inadequate CPV data collection and analysis capabilities compromise the analytical rigor that Section 14 demands.
The Interdependence of System Validation and Process Validation
Modern CPV programs depend entirely on computerized systems for data collection, statistical analysis, trend detection, and regulatory reporting. Manufacturing Execution Systems (MES) capture Critical Process Parameters (CPPs) in real-time. Laboratory Information Management Systems (LIMS) manage Critical Quality Attribute (CQA) testing data. Statistical process control platforms perform the normality testing, capability analysis, and control chart generation that drive CPV decision-making. Enterprise quality management systems integrate CPV findings with broader quality management activities including CAPA, change control, and regulatory reporting.
Section 14’s requirement that computerized systems remain “fit for intended use and in a validated state” directly impacts CPV program effectiveness and regulatory defensibility. A manufacturing execution system that undergoes undocumented configuration changes might continue to collect process data while compromising data integrity in ways that invalidate statistical analysis. A LIMS system with inadequate change control might introduce calculation errors that render capability analyses meaningless. Statistical software with unvalidated updates might generate control charts based on flawed algorithms.
The twelve pillars of Section 14 periodic review map directly onto CPV program dependencies. Hardware and software changes affect data collection accuracy and statistical calculation reliability. Documentation changes impact procedural consistency and analytical methodology validity. Combined effects of multiple changes create cumulative risks to data integrity that traditional CPV monitoring might not detect. Undocumented changes represent blind spots where system degradation occurs without CPV program awareness.
Risk-Based Integration: Aligning System Criticality with Process Impact
The risk-based approach fundamental to both CPV methodology and Section 14 periodic review creates opportunities for integrated assessment that optimizes resource allocation while ensuring comprehensive coverage. Systems supporting high-impact CPV parameters require more frequent and rigorous periodic review than those managing low-risk process monitoring.
Consider an example of a high-capability parameter with data clustered near LOQ requiring threshold-based alerts rather than traditional control charts. The computerized systems supporting this simplified monitoring approach—perhaps basic trending software with binary alarm capabilities—represent lower validation risk than sophisticated statistical process control platforms. Section 14’s risk-based frequency determination should reflect this reduced complexity, potentially extending review cycles while maintaining adequate oversight.
Conversely, systems supporting critical CPV parameters with complex statistical requirements—such as multivariate analysis platforms monitoring bioprocess parameters—warrant intensive periodic review given their direct impact on patient safety and product quality. These systems require comprehensive assessment of all twelve pillars with particular attention to change management, analytical method validation, and performance monitoring.
The integration extends to tool selection methodologies outlined in the CPV framework. Just as process parameters require different statistical tools based on data characteristics and risk profiles, the computerized systems supporting these tools require different validation and periodic review approaches. A system supporting simple attribute-based monitoring requires different periodic review depth than one performing sophisticated multivariate statistical analysis.
Data Integrity Convergence: CPV Analytics and System Audit Trails
Section 14’s emphasis on audit trail reviews and access reviews creates direct synergies with CPV data integrity requirements. The sophisticated statistical analyses required for effective CPV—including normality testing, capability analysis, and trend detection—depend on complete, accurate, and unaltered data throughout collection, storage, and analysis processes.
The framework’s discussion of decoupling analytical variability from process signals requires systems capable of maintaining separate data streams with independent validation and audit trail management. Section 14’s requirement to assess audit trail review effectiveness directly supports this CPV capability by ensuring that system-generated data remains traceable and trustworthy throughout complex analytical workflows.
Consider the example where threshold-based alerts replaced control charts for parameters near LOQ. This transition requires system modifications to implement binary logic, configure alert thresholds, and generate appropriate notifications. Section 14’s focus on combined effects of multiple changes ensures that such CPV-driven system modifications receive appropriate validation attention while the audit trail requirements ensure that the transition maintains data integrity throughout implementation.
The integration becomes particularly important for organizations implementing AI-enhanced CPV tools or advanced analytics platforms. These systems require sophisticated audit trail capabilities to maintain transparency in algorithmic decision-making while Section 14’s periodic review requirements ensure that AI model updates, training data changes, and algorithmic modifications receive appropriate validation oversight.
Living Risk Assessments: Dynamic Integration of System and Process Intelligence
The framework’s emphasis on living risk assessments that integrate ongoing data with periodic review cycles aligns perfectly with Section 14’s lifecycle approach to system validation. CPV programs generate continuous intelligence about process performance, parameter behavior, and statistical tool effectiveness that directly informs system validation decisions.
Process capability changes detected through CPV monitoring might indicate system performance degradation requiring investigation through Section 14 periodic review. Statistical tool effectiveness assessments conducted as part of CPV methodology might reveal system limitations requiring configuration changes or software updates. Risk profile evolution identified through living risk assessments might necessitate changes to Section 14 periodic review frequency or scope.
This dynamic integration creates feedback loops where CPV findings drive system validation decisions while system validation ensures CPV data integrity. Organizations must establish governance structures that facilitate information flow between CPV teams and system validation functions while maintaining appropriate independence in decision-making processes.
Implementation Framework: Integrating Section 14 with CPV Excellence
Organizations implementing both sophisticated CPV programs and Section 14 compliance should develop integrated governance frameworks that leverage synergies while avoiding duplication or conflicts. This requires coordinated planning that aligns system validation cycles with process validation activities while ensuring both programs receive adequate resources and management attention.
The implementation should begin with comprehensive mapping of system dependencies across CPV programs, identifying which computerized systems support which CPV parameters and analytical methods. This mapping drives risk-based prioritization of Section 14 periodic review activities while ensuring that high-impact CPV systems receive appropriate validation attention.
System validation planning should incorporate CPV methodology requirements including statistical software validation, data integrity controls, and analytical method computerization. CPV tool selection decisions should consider system validation implications including ongoing maintenance requirements, change control complexity, and periodic review resource needs.
Training programs should address the intersection of system validation and process validation requirements, ensuring that personnel understand both CPV statistical methodologies and computerized system compliance obligations. Cross-functional teams should include both process validation experts and system validation specialists to ensure decisions consider both perspectives.
Strategic Advantage Through Integration
Organizations that successfully integrate Section 14 system intelligence with CPV process intelligence will gain significant competitive advantages through enhanced decision-making capabilities, reduced compliance costs, and superior operational effectiveness. The combination creates comprehensive understanding of both process and system performance that enables proactive identification of risks and opportunities.
Integrated programs reduce resource requirements through coordinated planning and shared analytical capabilities while improving decision quality through comprehensive risk assessment and performance monitoring. Organizations can leverage system validation investments to enhance CPV capabilities while using CPV insights to optimize system validation resource allocation.
The integration also creates opportunities for enhanced regulatory relationships through demonstration of sophisticated compliance capabilities and proactive risk management. Regulatory agencies increasingly expect pharmaceutical organizations to leverage digital technologies for enhanced quality management, and the integration of Section 14 with CPV methodology demonstrates commitment to digital excellence and continuous improvement.
This integration represents the future of pharmaceutical quality management where system validation and process validation converge to create comprehensive intelligence systems that ensure product quality, patient safety, and regulatory compliance through sophisticated, risk-based, and continuously adaptive approaches. Organizations that master this integration will define industry best practices while building sustainable competitive advantages through operational excellence and regulatory sophistication.
In the annals of pharmaceutical regulation, few acronyms have generated as much discussion, confusion, and controversy as ALCOA. What began as a simple mnemonic device for FDA inspectors in the 1990s has evolved into a complex framework that has sparked heated debates across regulatory agencies, industry associations, and boardrooms worldwide. The story of ALCOA’s evolution from a five-letter inspector’s tool to the comprehensive ALCOA++ framework represents one of the most significant regulatory harmonization challenges of the modern pharmaceutical era.
With the publication of Draft EU GMP Chapter 4 in 2025, this three-decade saga of definitional disputes, regulatory inconsistencies, and industry resistance finally reaches its definitive conclusion. For the first time in regulatory history, a major jurisdiction has provided comprehensive, legally binding definitions for all ten ALCOA++ principles, effectively ending years of interpretive debates and establishing the global gold standard for pharmaceutical data integrity.
The Genesis: Stan Woollen’s Simple Solution
The ALCOA story begins in the early 1990s with Stan W. Woollen, an FDA inspector working in the Office of Enforcement. Faced with the challenge of training fellow GLP inspectors on data quality assessment, Woollen needed a memorable framework that could be easily applied during inspections. Drawing inspiration from the ubiquitous aluminum foil manufacturer, he created the ALCOA acronym: Attributable, Legible, Contemporaneous, Original, and Accurate.
“The ALCOA acronym was first coined by me while serving in FDA’s Office of Enforcement back in the early 1990’s,” Woollen later wrote in a 2010 retrospective. “Exactly when I first used the acronym I don’t recall, but it was a simple tool to help inspectors evaluate data quality”.
Woollen’s original intent was modest—create a practical checklist for GLP inspections. He explicitly noted that “the individual elements of ALCOA were already present in existing Good Manufacturing Practice (GMP) and GLP regulations. What he did was organize them into an easily memorized acronym”. This simple organizational tool would eventually become the foundation for a global regulatory framework.
The First Expansion: EMA’s ALCOA+ Revolution
The pharmaceutical landscape of 2010 bore little resemblance to Woollen’s 1990s GLP world. Electronic systems had proliferated, global supply chains had emerged, and data integrity violations were making headlines. Recognizing that the original five ALCOA principles, while foundational, were insufficient for modern pharmaceutical operations, the European Medicines Agency took a bold step.
In their 2010 “Reflection paper on expectations for electronic source data and data transcribed to electronic data collection tools in clinical trials,” the EMA introduced four additional principles: Complete, Consistent, Enduring, and Available—creating ALCOA+. This expansion represented the first major regulatory enhancement to Woollen’s original framework and immediately sparked industry controversy.
The Industry Backlash
The pharmaceutical industry’s response to ALCOA+ was swift and largely negative. Trade associations argued that the original five principles were sufficient and that additional requirements represented regulatory overreach. “The industry argued that the original 5 were sufficient; regulators needed modern additions,” as contemporary accounts noted.
The resistance wasn’t merely philosophical—it was economic. Each new principle required system validations, process redesigns, and staff retraining. For companies operating legacy paper-based systems, the “Enduring” and “Available” requirements posed particular challenges, often necessitating expensive digitization projects.
The Fragmentation: Regulatory Babel
What followed ALCOA+’s introduction was a period of regulatory fragmentation that would plague the industry for over a decade. Different agencies adopted different interpretations, creating a compliance nightmare for multinational pharmaceutical companies.
FDA’s Conservative Approach
The FDA, despite being the birthplace of ALCOA, initially resisted the European additions. Their 2016 “Data Integrity and Compliance with CGMP Guidance for Industry” focused primarily on the original five ALCOA principles, with only implicit references to the additional requirements8. This created a transatlantic divide where companies faced different standards depending on their regulatory jurisdiction.
MHRA’s Independent Path
The UK’s MHRA further complicated matters by developing their own interpretations in their 2018 “GxP Data Integrity Guidance.” While generally supportive of ALCOA+, the MHRA included unique provisions such as their emphasis on “permanent and understandable” under “legible,” creating yet another variant.
WHO’s Evolving Position
The World Health Organization initially provided excellent guidance in their 2016 document, which included comprehensive ALCOA explanations in Appendix 1. However, their 2021 revision removed much of this detail.
PIC/S Harmonization Attempt
The Pharmaceutical Inspection Co-operation Scheme (PIC/S) attempted to bridge these differences with their 2021 “Guidance on Data Integrity,” which formally adopted ALCOA+ principles. However, even this harmonization effort failed to resolve fundamental definitional inconsistencies between agencies.
The Traceability Controversy: ALCOA++ Emerges
Just as the industry began adapting to ALCOA+, European regulators introduced another disruption. The EMA’s 2023 “Guideline on computerised systems and electronic data in clinical trials” added a tenth principle: Traceability, creating ALCOA++.
The Redundancy Debate
The addition of Traceability sparked the most intense regulatory debate in ALCOA’s history. Industry experts argued that traceability was already implicit in the original ALCOA principles. As R.D. McDowall noted in Spectroscopy Online, “Many would argue that the criterion ‘traceable’ is implicit in ALCOA and ALCOA+. However, the implication of the term is the problem; it is always better in data regulatory guidance to be explicit”.
The debate wasn’t merely academic. Companies that had invested millions in ALCOA+ compliance now faced another round of system upgrades and validations. The terminology confusion was equally problematic—some agencies used ALCOA++, others preferred ALCOA+ with implied traceability, and still others created their own variants like ALCOACCEA.
Industry Frustration
By 2023, industry frustration had reached a breaking point. Pharmaceutical executives complained about “multiple naming conventions (ALCOA+, ALCOA++, ALCOACCEA) created market confusion”. Quality professionals struggled to determine which version applied to their operations, leading to over-engineering in some cases and compliance gaps in others.
The regulatory inconsistencies created particular challenges for multinational companies. A facility manufacturing for both US and European markets might need to maintain different data integrity standards for the same product, depending on the intended market—an operationally complex and expensive proposition.
The Global Harmonization Failure
Despite multiple attempts at harmonization through ICH, PIC/S, and bilateral agreements, the regulatory community failed to establish a unified ALCOA standard. Each agency maintained sovereign authority over their interpretations, leading to:
Definitional Inconsistencies: The same ALCOA principle had different definitions across agencies. “Attributable” might emphasize individual identification in one jurisdiction while focusing on system traceability in another.
Technology-Specific Variations: Some agencies provided technology-neutral guidance while others specified different requirements for paper versus electronic systems.
Enforcement Variations: Inspection findings varied significantly between agencies, with some inspectors focusing on traditional ALCOA elements while others emphasized ALCOA+ additions.
Economic Inefficiencies: Companies faced redundant validation efforts, multiple audit preparations, and inconsistent training requirements across their global operations.
Draft EU Chapter 4: The Definitive Resolution
Against this backdrop of regulatory fragmentation and industry frustration, the European Commission’s Draft EU GMP Chapter 4 represents a watershed moment in pharmaceutical regulation. For the first time in ALCOA’s three-decade history, a major regulatory jurisdiction has provided comprehensive, legally binding definitions for all ten ALCOA++ principles.
Comprehensive Definitions
The draft chapter doesn’t merely list the ALCOA++ principles—it provides detailed, unambiguous definitions for each. The “Attributable” definition spans multiple sentences, covering not just identity but also timing, change control, and system attribution. The “Legible” definition explicitly addresses dynamic data and search capabilities, resolving years of debate about electronic system requirements.
Technology Integration
Unlike previous guidance documents that treated paper and electronic systems separately, Chapter 4 provides unified definitions that apply regardless of technology. The “Original” definition explicitly addresses both static (paper) and dynamic (electronic) data, stating that “Information that is originally captured in a dynamic state should remain available in that state”.
Risk-Based Framework
The draft integrates ALCOA++ principles into a broader risk-based data governance framework, addressing long-standing industry concerns about proportional implementation. The risk-based approach considers both data criticality and data risk, allowing companies to tailor their ALCOA++ implementations accordingly.
Hybrid System Recognition
Acknowledging the reality of modern pharmaceutical operations, the draft provides specific guidance for hybrid systems that combine paper and electronic elements—a practical consideration absent from earlier ALCOA guidance.
The End of Regulatory Babel
Draft Chapter 4’s comprehensive approach should effectively ends the definitional debates that have plagued ALCOA implementation for over a decade. By providing detailed, legally binding definitions, the EU has created the global gold standard that other agencies will likely adopt or reference.
Global Influence
The EU’s pharmaceutical market represents approximately 20% of global pharmaceutical sales, making compliance with EU standards essential for most major manufacturers. When EU GMP requirements are updated, they typically influence global practices due to the market’s size and regulatory sophistication.
Regulatory Convergence
Early indications suggest other agencies are already referencing the EU’s ALCOA++ definitions in their guidance development. The comprehensive nature of Chapter 4’s definitions makes them attractive references for agencies seeking to update their own data integrity requirements.
Industry Relief
For pharmaceutical companies, Chapter 4 represents regulatory clarity after years of uncertainty. Companies can now design global data integrity programs based on the EU’s comprehensive definitions, confident that they meet or exceed requirements in other jurisdictions.
Lessons from the ALCOA Evolution
The three-decade evolution of ALCOA offers several important lessons for pharmaceutical regulation:
Organic Growth vs. Planned Development: ALCOA’s organic evolution from inspector tool to global standard demonstrates how regulatory frameworks can outgrow their original intent. The lack of coordinated development led to inconsistencies that persisted for years.
Industry-Regulatory Dialogue Importance: The most successful ALCOA developments occurred when regulators engaged extensively with industry. The EU’s consultation process for Chapter 4, while not without controversy, produced a more practical and comprehensive framework than previous unilateral developments.
Technology Evolution Impact: Each ALCOA expansion reflected technological changes in pharmaceutical manufacturing. The original principles addressed paper-based GLP labs, ALCOA+ addressed electronic clinical systems, and ALCOA++ addresses modern integrated manufacturing environments.
Global Harmonization Challenges: Despite good intentions, regulatory harmonization proved extremely difficult to achieve through international cooperation. The EU’s unilateral approach may prove more successful in creating de facto global standards.
The Future of Data Integrity
With Draft Chapter 4’s comprehensive ALCOA++ framework, the regulatory community has finally established a mature, detailed standard for pharmaceutical data integrity. The decades of debate, expansion, and controversy have culminated in a framework that addresses the full spectrum of modern pharmaceutical operations.
Implementation Timeline
The EU’s implementation timeline provides the industry with adequate preparation time while establishing clear deadlines for compliance. Companies have approximately 18-24 months to align their systems with the new requirements, allowing for systematic implementation without rushed remediation efforts.
Global Adoption
Early indications suggest rapid global adoption of the EU’s ALCOA++ definitions. Regulatory agencies worldwide are likely to reference or adopt these definitions in their own guidance updates, finally achieving the harmonization that eluded the international community for decades.
Technology Integration
The framework’s technology-neutral approach while addressing specific technology requirements positions it well for future technological developments. Whether dealing with artificial intelligence, blockchain, or yet-to-be-developed technologies, the comprehensive definitions provide a stable foundation for ongoing innovation.
Conclusion: From Chaos to Clarity
The evolution of ALCOA from Stan Woollen’s simple inspector tool to the comprehensive ALCOA++ framework represents one of the most significant regulatory development sagas in pharmaceutical history. Three decades of expansion, controversy, and fragmentation have finally culminated in the European Union’s definitive resolution through Draft Chapter 4.
For an industry that has struggled with regulatory inconsistencies, definitional debates, and implementation uncertainties, Chapter 4 represents more than just updated guidance—it represents regulatory maturity. The comprehensive definitions, risk-based approach, and technology integration provide the clarity that has been absent from data integrity requirements for over a decade.
The pharmaceutical industry can now move forward with confidence, implementing data integrity programs based on clear, comprehensive, and legally binding definitions. The era of ALCOA debates is over; the era of ALCOA++ implementation has begun.
As we look back on this regulatory journey, Stan Woollen’s simple aluminum foil-inspired acronym has evolved into something he likely never envisioned—a comprehensive framework for ensuring data integrity across the global pharmaceutical industry. The transformation from inspector’s tool to global standard demonstrates how regulatory innovation, while often messy and contentious, ultimately serves the critical goal of ensuring pharmaceutical product quality and patient safety.
The Draft EU Chapter 4 doesn’t just end the ALCOA debates—it establishes the foundation for the next generation of pharmaceutical data integrity requirements. For an industry built on evidence and data, having clear, comprehensive standards for data integrity represents a fundamental advancement in regulatory science and pharmaceutical quality assurance.