The GAO has published a report on FDA’s Inspections that found a 36% decrease compared to fiscal year 2019 in the number of inspections, partly due to reduced investigator capacity. A piece of information that should surprise noone.
The report highlights a concerning trend in the FDA’s drug inspection workforce. From November 2021 to June 2024, the vacancy rate among investigators who inspect foreign and domestic manufacturers increased from 9% to 16%.
I think we’ve all seen the impact of this. It’s worth spending a little time reading the report.
In the world of pharmaceutical manufacturing, cleanroom classifications play a crucial role in ensuring product quality and patient safety. However, a significant hurdle in the global harmonization of regulations has been a pain in our sides for a long time, that highlights the persistent differences between major regulatory bodies, including the FDA, EMA, and others, despite efforts to align through organizations like the World Health Organization (WHO) and the Pharmaceutical Inspection Co-operation Scheme (PIC/S).
The Current Landscape
United States Approach
In the United States, cleanroom classifications are primarily governed by two key documents:
The FDA’s “Sterile Drug Products Produced by Aseptic Processing” guidance
ISO 14644-1 standard for cleanroom classifications
The ISO 14644-1 standard is particularly noteworthy as it’s a general standard applicable across various industries utilizing cleanrooms, not just pharmaceuticals.
European Union Approach
The European Union takes a different stance, employing a grading system outlined in the EU GMP guide:
Grades A through D are used for normal cleanroom operation
ISO 14644 is still utilized, but primarily for validation purposes
World Health Organization Alignment
The World Health Organization (WHO) aligns with the European approach, adopting the same A to D grading system in its GMP guidelines.
The Implications of Disharmony
This lack of harmonization in cleanroom classifications presents several challenges:
Regulatory Complexity: Companies operating globally must navigate different classification systems, potentially leading to confusion and increased compliance costs.
Technology Transfer Issues: Transferring manufacturing processes between regions becomes more complicated when cleanroom requirements differ.
Inspection Inconsistencies: Differences in classification systems can lead to varying interpretations during inspections by different regulatory bodies.
The Missed Opportunity in Annex 1
The recent update to Annex 1, a key document in GMP regulations, could have been a prime opportunity to address this disharmony. However, despite involvement from WHO and PIC/S (and through them the FDA), the update failed to bring about the hoped-for alignment in cleanroom classifications.
Moving Forward
As the pharmaceutical industry continues to globalize, the need for harmonized regulations continues to be central. I would love to see future efforts towards harmonization here that would:
Prioritize alignment on fundamental technical specifications like cleanroom classifications
Consider the practical implications for manufacturers operating across multiple jurisdictions
While the journey towards full regulatory harmonization may be long and challenging, addressing key discrepancies like cleanroom classifications would represent a significant step forward for the global pharmaceutical industry.
FDA guidance, “Control of Nitrosamine Impurities in Human Drugs,” revises the final guidance of the same name issued on February 24, 2021, by including information about nitrosamine drug substance related impurities (NDSRIs), recommending implementation of new nitrosamine control strategies, and providing an updated timeline for manufacturers and applicants to implement these recommendations.
Nitrosamine impurities are important to control because they are potential human carcinogens. Long-term exposure to these impurities at levels above acceptable limits can increase the risk of cancer. Nitrosamines can be found in various consumer products and the environment, and they have been detected in several pharmaceutical products since 2018, prompting recalls and regulatory actions. A lot of regulatory action. Nitrosamine impurities may be one of the biggest drivers of changes in the GMPs.
Current Regulatory View
Regulators, including the FDA, Health Canada, and the European Medicines Agency (EMA), have been actively working to address the presence of nitrosamine impurities in medications. The current regulatory view emphasizes:
Risk Assessment and Control: Regulatory agencies have established acceptable intake (AI) limits for nitrosamines in drug products. These limits are designed to minimize the risk of cancer associated with long-term exposure to these impurities.
Guidance and Frameworks: Agencies have issued guidance documents outlining frameworks for assessing and controlling nitrosamine impurities. For example, the FDA’s guidance includes recommendations for predicting the mutagenic and carcinogenic potential of nitrosamine drug substance-related impurities (NDSRIs) and provides AI limits based on carcinogenic potency categorization.
International Collaboration: There is significant collaboration among global regulators to harmonize approaches and methodologies for controlling nitrosamine impurities. This includes the adoption of the Carcinogenic Potency Categorization Approach (CPCA) to determine AI limits.
Industry Responsibility: Manufacturers are responsible for understanding their processes to prevent nitrosamine formation and for conducting risk assessments. They must implement control strategies and perform confirmatory testing to ensure that nitrosamine levels remain below the established AI limits.
Regulators are focused on ensuring the safety of pharmaceutical products by controlling nitrosamine impurities through comprehensive risk assessments, setting stringent AI limits, and fostering international cooperation. Companies need to make sure they are ahead of this matter.
I hasn’t been difficult to notice that a whole lot of biological new drug applications have been rejected in the last few years, many for CMC reasons. Recently CDER Director Patrizia Cavazzoni spoke on the matter at a recent at a Duke University and FDA event at the National Press Club iin the video above.
“Our standards have not changed. We have exactly the same standards as we had in 2018 and 2019,” she said, before going on to talk about how the quality related issues the FDA is seeing: contamination, overall oversight, manufacturing controls or insufficient quality management systems.
Max Van Tassell, a senior pharmaceutical quality assessor in CDER’s Office of Pharmaceutical Quality, provided insights from analyzing 100 complete response letters (CRLs) for Biologics License Applications (BLAs) issued between 2014 and 2024. He noted that facility-related deficiencies in CRLs typically stem from inadequate demonstration that proposed corrective and preventive actions would effectively mitigate risks identified during on-site inspections.
It should be a key takeaway from this presentation that:
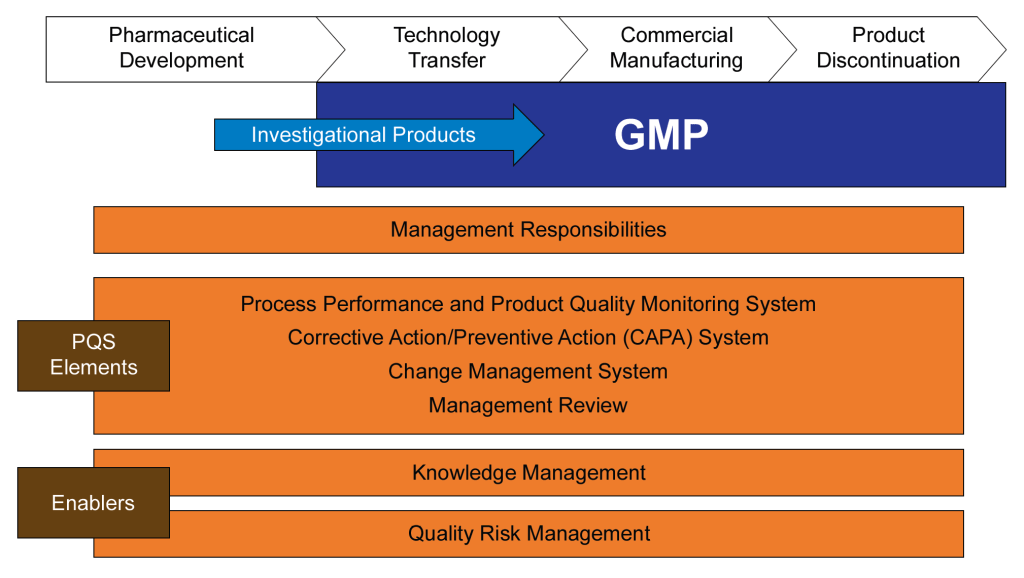
The International Conference on Harmonization (ICH) was established to harmonize the technical requirements for pharmaceutical product registration across Europe, Japan, and the United States. ICH Q10, finalized in June 2008, emerged from this initiative as a guideline for a comprehensive Pharmaceutical Quality System (PQS) applicable throughout the product lifecycle. It was adopted by the FDA in April 2009, following its implementation by the European Commission in July 2008.
ICH Q10 aims to provide a model for pharmaceutical manufacturers to develop and maintain effective quality management systems. The guideline emphasizes a lifecycle approach, integrating quality management principles from ISO standards and regional GMP requirements. The primary objectives of ICH Q10 include:
Ensuring consistent product quality that meets customer and regulatory requirements.
Establishing effective monitoring and control systems for process performance and product quality.
Promoting continual improvement and innovation throughout the product lifecycle.
The guideline outlines the key elements of management responsibilities, Corrective and Preventive Action (CAPA) , process performance and product quality monitoring, change management, and management review. ICH Q10 is usually considered part of the “Quality Trio” with ICH Q8 and Q9. Quality by design is only possible through proper risk management and a robust quality system.
FDA Guidance for Industry on Quality Systems Approach to Pharmaceutical CGMP Regulation
The FDA developed guidance on implementing modern quality systems and risk management practices to align with the CGMP (Current Good Manufacturing Practice) requirements outlined in parts 210 and 211 of the FDA regulations. These regulations govern the manufacturing of human and veterinary drugs, including biological products. Published in 2006, this guidance should be viewed as part of a continuum of thought with ICH Q10 and not as an earlier draft.
This guidance aims to assist manufacturers in meeting cGMP requirements by adopting a comprehensive quality systems model. It emphasizes the integration of quality systems with regulatory requirements to ensure full compliance without imposing new expectations on manufacturers. Key aspects of the guidance include:
Highlighting the consistency of the quality systems model with cGMP regulations.
Encouraging the use of risk management and quality systems to enhance compliance and product quality.
Providing a framework for manufacturers to gain control over their manufacturing processes.
Six-System Inspection Model
The FDA’s Six-System Inspection Model is a framework introduced in this guidance to ensure compliance with current Good Manufacturing Practice (CGMP) regulations in the pharmaceutical industry. This model helps FDA inspectors evaluate the robustness of a company’s quality management system by focusing on six key subsystems.
I am a huge fan of the six subsystem approach. Basically we have here the organization of the quality manual, a guide to what standards you need to write in a bigger company, and a franework for understanding the cGMPs as a whole (great for education purposes).
Here’s a detailed explanation of each subsystem:
1. Quality System
Role: Acts as the central hub for all other systems, ensuring overall quality management.
Focus: Management responsibilities, internal audits, CAPA (Corrective and Preventive Actions), and continuous improvement.
Importance: Ensures that all other systems are effectively integrated and managed to maintain product quality and regulatory compliance.
2. Facilities and Equipment System
Role: Ensures that facilities and equipment are suitable for their intended use and maintained properly.
Focus: Design, maintenance, cleaning, and calibration of facilities and equipment.
Importance: Prevents contamination and ensures consistent manufacturing conditions.
3. Materials System
Role: Manages the control of raw materials, components, and packaging materials.
Focus: Supplier qualification, receipt, storage, inventory control, and testing of materials.
Importance: Ensures that only high-quality materials are used in the manufacturing process, reducing the risk of product defects.
4. Production System
Role: Oversees the actual manufacturing processes.
Focus: Process controls, batch records, in-process controls, and validation.
Importance: Ensures that products are manufactured consistently and meet predefined quality criteria.
5. Packaging and Labeling System
Role: Manages the packaging and labeling processes to ensure correct and compliant product presentation.
Focus: Label control, packaging operations, and labeling verification.
Importance: Prevents mix-ups and ensures that products are correctly identified and used.
6. Laboratory Controls System
Role: Ensures the reliability of laboratory testing and data integrity.
Focus: Sampling, testing, analytical method validation, and laboratory records.
Importance: Verifies that products meet quality specifications before release.
Integration and Interdependence
Quality System as the Fulcrum: The quality system is the central element that integrates all other subsystems. It ensures that each subsystem functions correctly and is aligned with overall quality objectives.
State of Control: The primary goal of the six-system inspection model is to ensure that each subsystem is in a state of control, meaning it operates within predefined limits and consistently produces the desired outcomes.
The Six-System Inspection Model provides a structured approach for FDA inspectors to assess the compliance and effectiveness of a pharmaceutical company’s quality management system. By focusing on these six subsystems, the FDA ensures that all aspects of manufacturing, from raw materials to final product testing, are adequately controlled and managed to maintain high standards of product quality and safety.
A Complementary and Holistic Approach
Both ICH Q10 and the FDA’s guidance on quality systems approach aim to enhance the quality and safety of pharmaceutical products through robust quality management systems. ICH Q10 provides a harmonized model applicable across the product lifecycle, while the FDA guidance focuses on integrating quality systems with existing CGMP regulations. Together, they support the pharmaceutical industry in achieving consistent product quality and regulatory compliance.
Aspect
ICH Q10
FDA Guidance on CGMP
ISO 13485 and 21 CFR 820
ISO 9000
Purpose and Scope
Comprehensive model for pharmaceutical quality systems across the product lifecycle.
Quality systems approach to ensure CGMP compliance in pharmaceuticals.
Quality management system for medical devices, incorporating ISO 13485 and regulatory requirements of 21 CFR 820.
Fundamentals and vocabulary for quality management systems applicable to any industry.
Industry Focus
Specifically for the pharmaceutical industry.
Specifically for the pharmaceutical industry.
Specifically for the medical device industry.
Applicable to any industry.
Key Elements
Management responsibilities, CAPA, process performance, change management, management review.
Management responsibilities, quality systems, process validation, continuous improvement.
Quality management principles, terms, and definitions.
Regulatory Focus
Strong emphasis on regulatory compliance and lifecycle management.
Strong emphasis on regulatory compliance with CGMP.
Incorporates regulatory requirements specific to medical devices (21 CFR 820).
Does not directly address regulatory compliance.
Flexibility
Flexible, adaptable to specific product and process needs.
More prescriptive with specific compliance requirements.
Harmonized with international standards but includes specific regulatory requirements.
Provides a broad framework for customization.
Management Involvement
Emphasizes management’s role in quality and regulatory compliance.
Emphasizes management’s role in quality and CGMP integration.
Emphasizes management’s role in quality and risk-based decision making.
Emphasizes management’s role in quality and customer satisfaction.
Implementation
Tailored to pharmaceutical manufacturing, integrating quality management principles.
Mandates oversight and controls over drug manufacturing processes.
Requires a quality manual and specific documentation practices; aligned with international standards.
Requires customization to specific industry needs.
These two documents were developed at the same time and represents the thinking twenty years ago in laying down an approach that still matters today. I usually regard the six system approach as a deepening and defining of what Q10 means by process performance and product quality monitoring.
What is the current agency thinking?
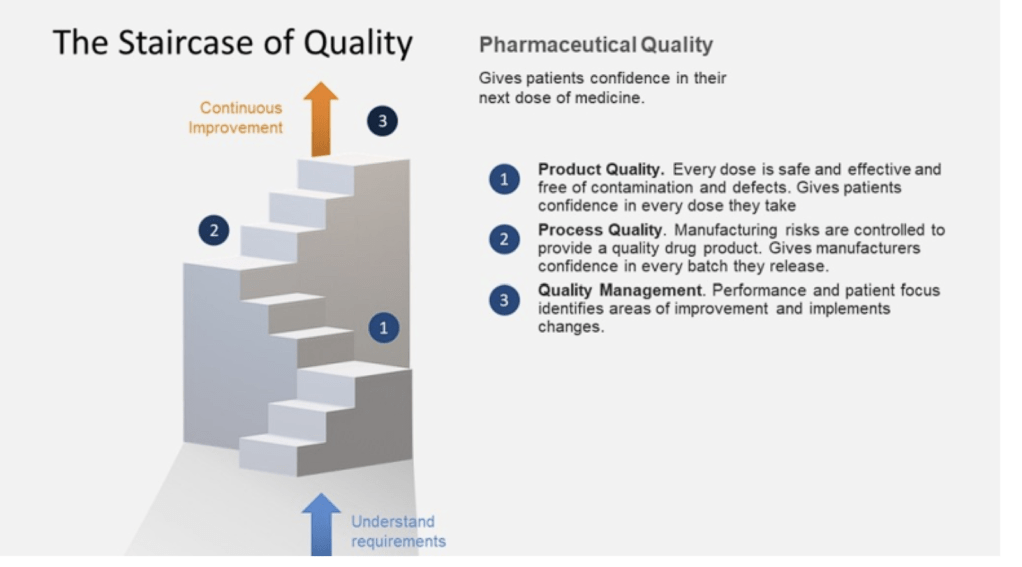
The FDA and other revulatory agencies haven’t stopped their thinking in 2008. Sixteen years later we see the continued push for quality culture and quality maturity. The FDA continues to make this a top priority, as we’ve been seeing in their annual drug shortage reports to Congress. There are a few themes we continue to see driven home.
The Patient is the Customer
Quality management must be customer-focused, ensuring that all processes and materials meet their intended use. Senior management’s commitment is crucial for a strong QMS, which emphasizes proactive quality assurance over reactive quality control. Robust supplier relationships and oversight programs are essential to manage variability in materials and processes.
This application of a core priciple in ISO 9000 may seem to basic to some, but I think it is central to a lot of messaging and should never be taken for granted.
Benefits of Better Quality Performance
A continued focus that a quality-focused culture leads to:
Early problem detection
Enhanced process stability and productivity
Fewer major deviations and failures
Efficient QA release of batches
Reduced customer complaints and returns
Protection of brand and competitiveness
Management Oversight of Drug Quality
Management must address sources of variability, including people, materials, methods, measurements, machines, and environment. Risk management should be dynamic and ongoing, facilitating continual learning and improvement.
Corrective Action and Preventive Action (CAPA)
A structured approach to investigating complaints, product rejections, nonconformances, recalls, deviations, audits, regulatory inspections, and trends is essential. CAPA should determine root causes and implement corrective actions.
Change Management
Timely and effective change management ensures corrections and improvements are undertaken efficiently. This includes implementing product quality improvements, process improvements, variability reduction, innovations, and pharmaceutical quality system enhancements.
Management Review
Management is responsible for quality policy, QMS effectiveness, internal communications, resource management, and supply chain oversight. This includes ensuring the quality of incoming materials and outsourced activities.
Quality Culture Driven by Top Management
A strong corporate quality culture is driven by daily decisions and executive oversight. Sustainable compliance requires aiming for high standards rather than just meeting minimum requirements. Quality management maturity involves proactive and preventive actions, iterative learning, and leveraging modern technologies.
Facility Lifecycle
Senior management must ensure the suitability of operational design, control, and maintenance. This includes addressing infrastructure reliability, appropriateness for new product demands, and mitigating equipment/facility degradation.
Risk Management in Manufacturing
Human factors and manual interventions pose significant risks in pharmaceutical manufacturing. Automation and separation technologies can mitigate these risks, but many facilities still rely on manually intensive processes. Leveraging new technologies and practices is a huge opportunity.
This approach is reflected in the FDA’s Quality Management Maturity (QMM), which promotes advanced quality management practices within drug manufacturing establishments.
Goals of the QMM Program
Foster a Strong Quality Culture Mindset: Encourage establishments to integrate quality deeply into their organizational culture.
Recognize Advanced Quality Management Practices: Acknowledge and reward establishments that go beyond basic CGMP (Current Good Manufacturing Practices) requirements.
Identify Growth Opportunities: Provide suggestions for enhancing quality management practices.
Minimize Risks to Product Availability: Ensure a reliable market supply by reducing quality-related failures and maintaining performance during supply chain disruptions.
Key Components of the QMM Program
Management Commitment to Quality: Leadership must prioritize quality, set clear objectives, and integrate these with business goals. Effective management review processes are crucial.
Business Continuity: Establishments should develop robust plans to handle disruptions, ensuring consistent operations and supply chain reliability.
Advanced Pharmaceutical Quality System (PQS): Implementing quality principles like Quality by Design (QbD) and risk management approaches to maintain system reliability and minimize production disruptions.
Technical Excellence: Emphasizing data management, innovative manufacturing processes, and advanced technologies to enhance quality and operational efficiency.
Employee Engagement and Empowerment: Encouraging employees to take ownership of quality, make suggestions, and understand their impact on product quality and patient safety.
Implementation and Assessment
The FDA has developed a prototype assessment protocol to evaluate QMM. This includes a standardized approach to minimize bias and ensure objectivity. Someday, eventually, it will move away from constant prototyping.
Assessments will focus on qualitative aspects, such as the establishment’s quality culture and how it uses data to drive improvements.
Benefits of QMM
Enhanced Supply Chain Reliability: By adopting mature quality management practices, establishments can reduce the occurrence of quality-related failures. The fact shortages continue to be so damning to our industry is a huge wake-up call.
Proactive Continual Improvement: Encourages a proactive approach to quality management, leveraging technological advancements and integrated business operations.
Long-term Cost Savings: Investing in a mature quality culture can lead to fewer compliance issues, reduced inspection needs, and overall cost reductions.
Conclusion
The FDA’s QMM program aims to transform how pharmaceutical quality is perceived, measured, and rewarded. The program seeks to ensure a more reliable drug supply and better patient outcomes by fostering a strong quality culture and recognizing advanced practices. It should be seen as part of a 20-year commitment from the agency in alignment with its international partners.