“there is no retrospective review of batch records for batches within expiry, to identify any other process deviations performed without the appropriate corresponding documentation including risk assessment(s).” – 2025 Warning Letter from the US FDA to Sanofi
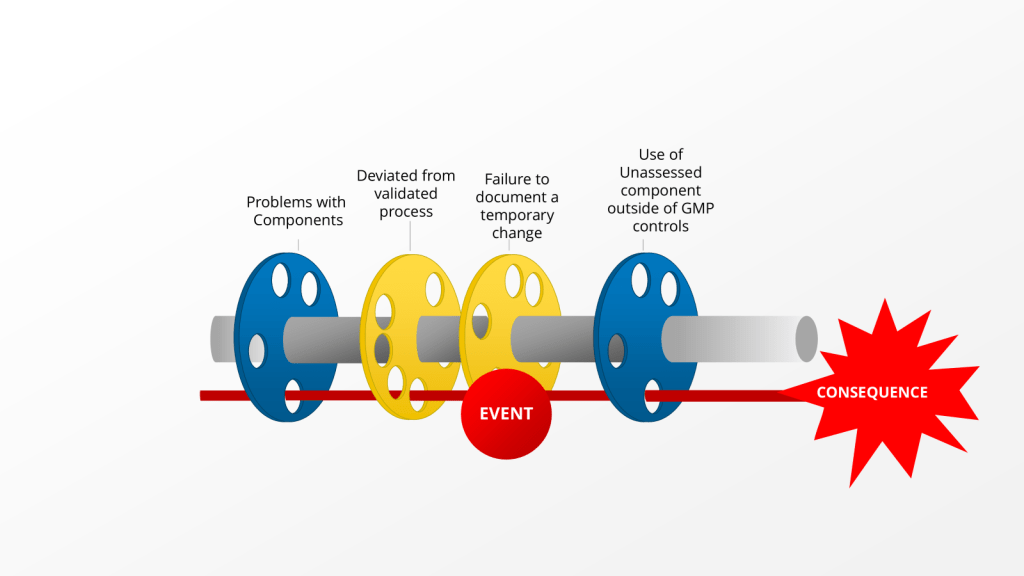
This comment is about an instance where Sanofi deviated from the validated process by using an unvalidated single use component. Instead of self-identifying, creating a deviation and doing the right change control activities, the company just kept on deviating by using a non-controlled document.
This is a big problem for lots of reasons, from uncontrolled documents, to not using the change control system, to breaking the validated state. What the language quoted above really brings to bear is the question, when should we evaluate our records for other similar instances of this happening, so we can address it.
When a deviation investigation reveals recurring bad decision-making, it is crucial to expand the investigation and conduct a retrospective review of batch records. A good cutoff of this can be only for batches within expiry. This expanded investigation helps identify any other process deviations that may have occurred but were not discovered or documented at the time. Here’s when and how to approach this situation:
Triggers for Expanding the Investigation
- Recurring Deviations: If the same or similar deviations are found to be recurring, it indicates a systemic issue that requires a broader investigation.
- Pattern of Human Errors: When a pattern of human errors or poor decision-making is identified, it suggests potential underlying issues in training, procedures, or processes.
- Critical Deviations: For deviations classified as critical, a more thorough investigation is typically warranted, including a retrospective review.
- Potential Impact on Product Quality: If there’s a strong possibility that undiscovered deviations could affect product quality or patient safety, an expanded investigation becomes necessary.
Conducting the Retrospective Review
- Timeframe: Review batch records for all batches within expiry, typically covering at least two years of production. Similarily for issues in the FUSE program you might look since the last requalification, or from a decide to go backwards in concentric circles based on what you find.
- Scope: Examine not only the specific process where the deviation was found but also related processes or areas that could be affected. Reviewing related processes is critical.
- Data Analysis: Utilize statistical tools and trending analysis techniques to identify patterns or anomalies in the historical data.
- Cross-Functional Approach: Involve a team of subject matter experts from relevant departments to ensure a comprehensive review.
- Documentation Review: Examine batch production records, laboratory control records, equipment logs, and any other relevant documentation.
- Root Cause Analysis: Apply root cause analysis techniques to understand the underlying reasons for the recurring issues.
Key Considerations
- Risk Assessment: Prioritize the review based on the potential risk to product quality and patient safety.
- Data Integrity: Ensure that any retrospective data used is reliable and has maintained its integrity.
- Corrective Actions: Develop and implement corrective and preventive actions (CAPAs) based on the findings of the expanded investigation.
- Regulatory Reporting: Assess the need for notifying regulatory authorities based on the severity and impact of the findings.
By conducting a thorough retrospective review when recurring bad decision-making is identified, companies can uncover hidden issues, improve their quality systems, and prevent future deviations. This proactive approach not only enhances compliance but also contributes to continuous improvement in pharmaceutical manufacturing processes.
In the case of an issue that rises to a regulatory observation this becomes a firm must. The agency has raised a significant concern and they will want proof that this is a limited issue or that you are holistically dealing with it across the organization.
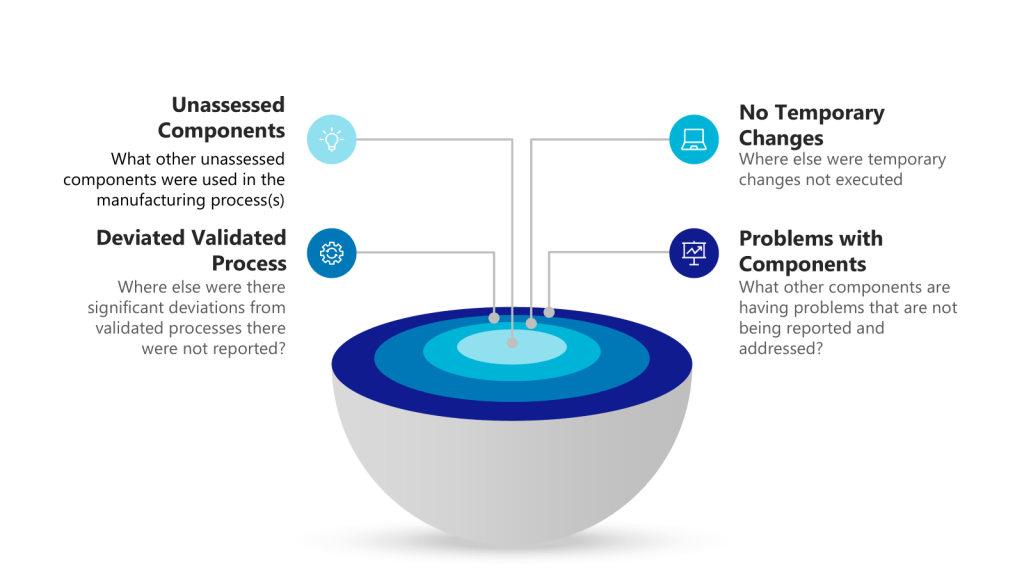
Concentric Circles of Investigation
Each layer of the investigation may require holistic looks. Utilizing the example above we have:
| Layer of Problem | Further Investigation to Answer |
| Use of unassessed component outside of GMP controls | What other unassessed components were used in the manufacturing process(s) |
| Failure to document a temporary change | Where else were temporary changes not executed |
| Deviated from validated process | Where else were there significant deviations from validated processes there were not reported |
| Problems with components | What other components are having problems that are not being reported and addressed |
Take a risk-based approach here is critical.