The Pfizer McPherson site has been under a great deal of regulatory scrutiny, and as a result there is a lot we can learn from their findings.
In July the MHRA stated the following:

There is a lot to unpack here, and for most of it I can pull up some previous postings to start with:
Breaking down change controls is both a necessity and a difficulty. I talked about the need for a change strategy when breaking up changes. This connective tissue will help with issues like 2.4.1.1 above and can also serve as a good playbook for discussing the changes with an inspector. This is especially important when you find you need to implement related changes at different times. I talked about the various implementation dates in some detail.
Risk assessments are only getting more important, and for a company with international distribution it is important to consider the risks inherent to your regulatory strategy and distribution strategy and mitigate.
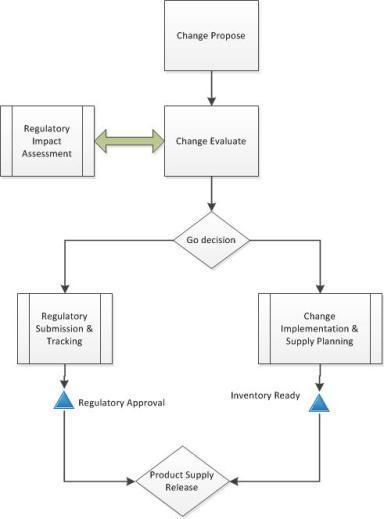
If you have changes that will have long tails of regulatory approvals, then your change control needs to have the right controls to ensure appropriate and safe supply.
Build your actions to address all risks and impacts and ensure they are appropriately carried through.
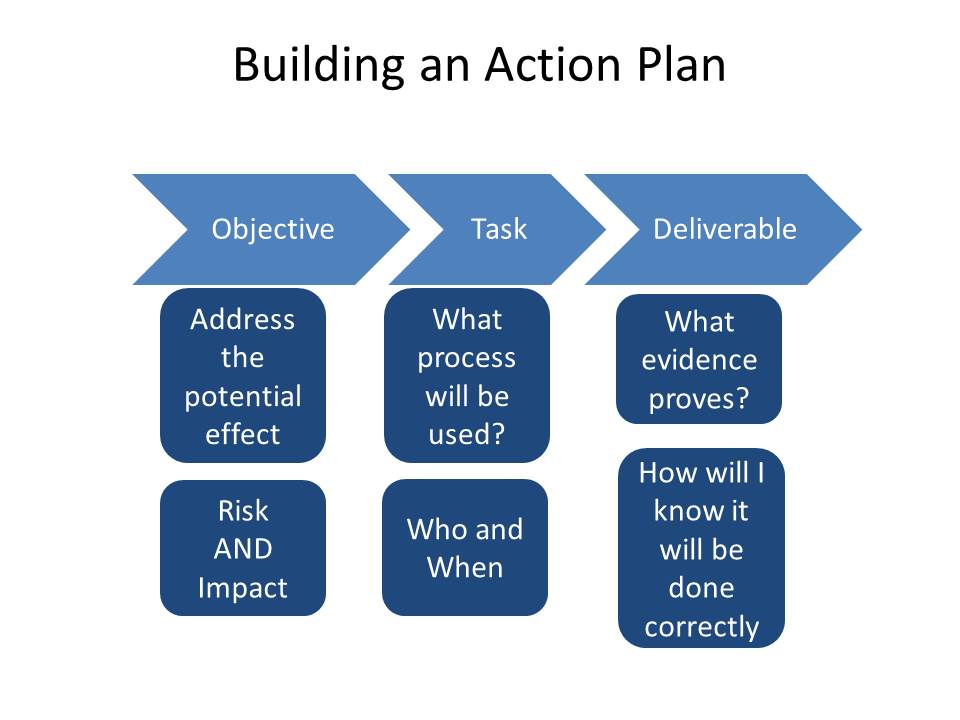
Finally ensure your change control process has a way to revise the plan and ensure all stakeholders are included in the decisions.
