As quality professionals, we can often fall into the trap of believing that more analysis, more data, and more complex decision trees lead to better outcomes. But what if this fundamental assumption is not just wrong, but actively harmful to effective risk management? Gerd Gigerenzer‘s decades of research on bounded rationality and fast-and-frugal heuristics suggests exactly that—and the implications for how we approach quality risk management are profound.
The Myth of Optimization in Risk Management
Too much of our risk management practice assumes we operate like Laplacian demons—omniscient beings with unlimited computational power and perfect information. Gigerenzer calls this “unbounded rationality,” and it’s about as realistic as expecting your quality management system to implement itself.
In reality, experts operate under severe constraints: limited time, incomplete information, constantly changing regulations, and the perpetual pressure to balance risk mitigation with operational efficiency. How we move beyond thinking of these as bugs to be overcome, and build tools that address these concerns is critical to thinking of risk management as a science.
Enter the Adaptive Toolbox
Gigerenzer’s adaptive toolbox concept revolutionizes how we think about decision-making under uncertainty. Rather than viewing our mental shortcuts (heuristics) as cognitive failures that need to be corrected, the adaptive toolbox framework recognizes them as evolved tools that can outperform complex analytical methods in real-world conditions.
The toolbox consists of three key components that every risk manager should understand:
Search Rules: How we look for information when making risk decisions. Instead of trying to gather all possible data (which is impossible anyway), effective heuristics use smart search strategies that focus on the most diagnostic information first.
Stopping Rules: When to stop gathering information and make a decision. This is crucial in quality management where analysis paralysis can be as dangerous as hasty decisions.
Decision Rules: How to integrate the limited information we’ve gathered into actionable decisions.
These components work together to create what Gigerenzer calls “ecological rationality”—decision strategies that are adapted to the specific environment in which they operate. For quality professionals, this means developing risk management approaches that fit the actual constraints and characteristics of pharmaceutical manufacturing, not the theoretical world of perfect information.
The Less-Is-More Revolution
One of Gigerenzer’s most counterintuitive findings is the “less-is-more effect”—situations where ignoring information actually leads to better decisions. This challenges everything we think we know about evidence-based decision making in quality.
Consider an example from emergency medicine that directly parallels quality risk management challenges. When patients arrive with chest pain, doctors traditionally used complex diagnostic algorithms considering up to 19 different risk factors. But researchers found that a simple three-question decision tree outperformed the complex analysis in both speed and accuracy.
The fast-and-frugal tree asked only:
Are there ST segment changes on the EKG?
Is chest pain the chief complaint?
Does the patient have any additional high-risk factors?
Based on these three questions, doctors could quickly and accurately classify patients as high-risk (requiring immediate intensive care) or low-risk (suitable for regular monitoring). The key insight: the simple approach was not just faster—it was more accurate than the complex alternative.
Applying Fast-and-Frugal Trees to Quality Risk Management
This same principle applies directly to quality risk management decisions. Too often, we create elaborate risk assessment matrices that obscure rather than illuminate the critical decision factors. Fast-and-frugal trees offer a more effective alternative.
Let’s consider deviation classification—a daily challenge for quality professionals. Instead of complex scoring systems that attempt to quantify every possible risk dimension, a fast-and-frugal tree might ask:
Does this deviation involve a patient safety risk? If yes → High priority investigation (exit to immediate action)
Does this deviation affect product quality attributes? If yes → Standard investigation timeline
Is this a repeat occurrence of a similar deviation? If yes → Expedited investigation, if no → Routine handling
This simple decision tree accomplishes several things that complex matrices struggle with. First, it prioritizes patient safety above all other considerations—a value judgment that gets lost in numerical scoring systems. Second, it focuses investigative resources where they’re most needed. Third, it’s transparent and easy to train staff on, reducing variability in risk classification.
The beauty of fast-and-frugal trees isn’t just their simplicity. It is their robustness. Unlike complex models that break down when assumptions are violated, simple heuristics tend to perform consistently across different conditions.
The Recognition Heuristic in Supplier Quality
Another powerful tool from Gigerenzer’s adaptive toolbox is the recognition heuristic. This suggests that when choosing between two alternatives where one is recognized and the other isn’t, the recognized option is often the better choice.
In supplier qualification decisions, quality professionals often struggle with elaborate vendor assessment schemes that attempt to quantify every aspect of supplier capability. But experienced quality professionals know that supplier reputation—essentially a form of recognition—is often the best predictor of future performance.
The recognition heuristic doesn’t mean choosing suppliers solely on name recognition. Instead, it means understanding that recognition reflects accumulated positive experiences across the industry. When coupled with basic qualification criteria, recognition can be a powerful risk mitigation tool that’s more robust than complex scoring algorithms.
This principle extends to regulatory decision-making as well. Experienced quality professionals develop intuitive responses to regulatory trends and inspector concerns that often outperform elaborate compliance matrices. This isn’t unprofessional—it’s ecological rationality in action.
Take-the-Best Heuristic for Root Cause Analysis
The take-the-best heuristic offers an alternative approach to traditional root cause analysis. Instead of trying to weight and combine multiple potential root causes, this heuristic focuses on identifying the single most diagnostic factor and basing decisions primarily on that information.
In practice, this might mean:
Identifying potential root causes in order of their diagnostic power
Investigating the most powerful indicator first
If that investigation provides a clear direction, implementing corrective action
Only continuing to secondary factors if the primary investigation is inconclusive
This approach doesn’t mean ignoring secondary factors entirely, but it prevents the common problem of developing corrective action plans that try to address every conceivable contributing factor, often resulting in resource dilution and implementation challenges.
Managing Uncertainty in Validation Decisions
Validation represents one of the most uncertainty-rich areas of quality management. Traditional approaches attempt to reduce uncertainty through exhaustive testing, but Gigerenzer’s work suggests that some uncertainty is irreducible—and that trying to eliminate it entirely can actually harm decision quality.
Consider computer system validation decisions. Teams often struggle with determining how much testing is “enough,” leading to endless debates about edge cases and theoretical scenarios. The adaptive toolbox approach suggests developing simple rules that balance thoroughness with practical constraints:
The Satisficing Rule: Test until system functionality meets predefined acceptance criteria across critical business processes, then stop. Don’t continue testing just because more testing is theoretically possible.
The Critical Path Rule: Focus validation effort on the processes that directly impact patient safety and product quality. Treat administrative functions with less intensive validation approaches.
The Experience Rule: Leverage institutional knowledge about similar systems to guide validation scope. Don’t start every validation from scratch.
These heuristics don’t eliminate validation rigor—they channel it more effectively by recognizing that perfect validation is impossible and that attempting it can actually increase risk by delaying system implementation or consuming resources needed elsewhere.
Ecological Rationality in Regulatory Strategy
Perhaps nowhere is the adaptive toolbox more relevant than in regulatory strategy. Regulatory environments are characterized by uncertainty, incomplete information, and time pressure—exactly the conditions where fast-and-frugal heuristics excel.
Successful regulatory professionals develop intuitive responses to regulatory trends that often outperform complex compliance matrices. They recognize patterns in regulatory communications, anticipate inspector concerns, and adapt their strategies based on limited but diagnostic information.
The key insight from Gigerenzer’s work is that these intuitive responses aren’t unprofessional—they represent sophisticated pattern recognition based on evolved cognitive mechanisms. The challenge for quality organizations is to capture and systematize these insights without destroying their adaptive flexibility.
This might involve developing simple decision rules for common regulatory scenarios:
The Precedent Rule: When facing ambiguous regulatory requirements, look for relevant precedent in previous inspections or industry guidance rather than attempting exhaustive regulatory interpretation.
The Proactive Communication Rule: When regulatory risk is identified, communicate early with authorities rather than developing elaborate justification documents internally.
The Materiality Rule: Focus regulatory attention on changes that meaningfully affect product quality or patient safety rather than attempting to address every theoretical concern.
Building Adaptive Capability in Quality Organizations
Implementing Gigerenzer’s insights requires more than just teaching people about heuristics—it requires creating organizational conditions that support ecological rationality. This means:
Embracing Uncertainty: Stop pretending that perfect risk assessments are possible. Instead, develop decision-making approaches that are robust under uncertainty.
Valuing Experience: Recognize that experienced professionals’ intuitive responses often reflect sophisticated pattern recognition. Don’t automatically override professional judgment with algorithmic approaches.
Simplifying Decision Structures: Replace complex matrices and scoring systems with simple decision trees that focus on the most diagnostic factors.
Encouraging Rapid Iteration: Rather than trying to perfect decisions before implementation, develop approaches that allow rapid adjustment based on feedback.
Training Pattern Recognition: Help staff develop the pattern recognition skills that support effective heuristic decision-making.
The Subjectivity Challenge
One common objection to heuristic-based approaches is that they introduce subjectivity into risk management decisions. This concern reflects a fundamental misunderstanding of both traditional analytical methods and heuristic approaches.
Traditional risk matrices and analytical methods appear objective but are actually filled with subjective judgments: how risks are defined, how probabilities are estimated, how impacts are categorized, and how different risk dimensions are weighted. These subjective elements are simply hidden behind numerical facades.
Heuristic approaches make subjectivity explicit rather than hiding it. This transparency actually supports better risk management by forcing teams to acknowledge and discuss their value judgments rather than pretending they don’t exist.
The recent revision of ICH Q9 explicitly recognizes this challenge, noting that subjectivity cannot be eliminated from risk management but can be managed through appropriate process design. Fast-and-frugal heuristics support this goal by making decision logic transparent and teachable.
Four Essential Books by Gigerenzer
For quality professionals who want to dive deeper into this framework, here are four books by Gigerenzer to read:
1. “Simple Heuristics That Make Us Smart” (1999) – This foundational work, authored with Peter Todd and the ABC Research Group, establishes the theoretical framework for the adaptive toolbox. It demonstrates through extensive research how simple heuristics can outperform complex analytical methods across diverse domains. For quality professionals, this book provides the scientific foundation for understanding why less can indeed be more in risk assessment.
2. “Gut Feelings: The Intelligence of the Unconscious” (2007) – This more accessible book explores how intuitive decision-making works and when it can be trusted. It’s particularly valuable for quality professionals who need to balance analytical rigor with practical decision-making under pressure. The book provides actionable insights for recognizing when to trust professional judgment and when more analysis is needed.
3. “Risk Savvy: How to Make Good Decisions” (2014) – This book directly addresses risk perception and management, making it immediately relevant to quality professionals. It challenges common misconceptions about risk communication and provides practical tools for making better decisions under uncertainty. The sections on medical decision-making are particularly relevant to pharmaceutical quality management.
4. “The Intelligence of Intuition” (Cambridge University Press, 2023) – Gigerenzer’s latest work directly challenges the widespread dismissal of intuitive decision-making in favor of algorithmic solutions. In this compelling analysis, he traces what he calls the “war on intuition” in social sciences, from early gendered perceptions that dismissed intuition as feminine and therefore inferior, to modern technological paternalism that argues human judgment should be replaced by perfect algorithms. For quality professionals, this book is essential reading because it demonstrates that intuition is not irrational caprice but rather “unconscious intelligence based on years of experience” that evolved specifically to handle uncertain and dynamic situations where logic and big data algorithms provide little benefit. The book provides both theoretical foundation and practical guidance for distinguishing reliable intuitive responses from wishful thinking—a crucial skill for quality professionals who must balance analytical rigor with rapid decision-making under uncertainty.
The Implementation Challenge
Understanding the adaptive toolbox conceptually is different from implementing it organizationally. Quality systems are notoriously resistant to change, particularly when that change challenges fundamental assumptions about how decisions should be made.
Successful implementation requires a gradual approach that demonstrates value rather than demanding wholesale replacement of existing methods. Consider starting with pilot applications in lower-risk areas where the benefits of simpler approaches can be demonstrated without compromising patient safety.
Phase 1: Recognition and Documentation – Begin by documenting the informal heuristics that experienced staff already use. You’ll likely find that your most effective team members already use something resembling fast-and-frugal decision trees for routine decisions.
Phase 2: Formalization and Testing – Convert informal heuristics into explicit decision rules and test them against historical decisions. This helps build confidence and identifies areas where refinement is needed.
Phase 3: Training and Standardization – Train staff on the formalized heuristics and create simple reference tools that support consistent application.
Phase 4: Continuous Adaptation – Build feedback mechanisms that allow heuristics to evolve as conditions change and new patterns emerge.
Measuring Success with Ecological Metrics
Traditional quality metrics often focus on process compliance rather than decision quality. Implementing an adaptive toolbox approach requires different measures of success.
Instead of measuring how thoroughly risk assessments are documented, consider measuring:
Decision Speed: How quickly can teams classify and respond to different types of quality events?
Decision Consistency: How much variability exists in how similar situations are handled?
Resource Efficiency: What percentage of effort goes to analysis versus action?
Adaptation Rate: How quickly do decision approaches evolve in response to new information?
Outcome Quality: What are the actual consequences of decisions made using heuristic approaches?
These metrics align better with the goals of effective risk management: making good decisions quickly and consistently under uncertainty.
The Training Implication
If we accept that heuristic decision-making is not just inevitable but often superior, it changes how we think about quality training. Instead of teaching people to override their intuitive responses with analytical methods, we should focus on calibrating and improving their pattern recognition abilities.
This means:
Case-Based Learning: Using historical examples to help staff recognize patterns and develop appropriate responses
Scenario Training: Practicing decision-making under time pressure and incomplete information
Feedback Loops: Creating systems that help staff learn from decision outcomes
Expert Mentoring: Pairing experienced professionals with newer staff to transfer tacit knowledge
Cross-Functional Exposure: Giving staff experience across different areas to broaden their pattern recognition base
Addressing the Regulatory Concern
One persistent concern about heuristic approaches is regulatory acceptability. Will inspectors accept fast-and-frugal decision trees in place of traditional risk matrices?
The key insight from Gigerenzer’s work is that regulators themselves use heuristics extensively in their inspection and decision-making processes. Experienced inspectors develop pattern recognition skills that allow them to quickly identify potential problems and focus their attention appropriately. They don’t systematically evaluate every aspect of a quality system—they use diagnostic shortcuts to guide their investigations.
Understanding this reality suggests that well-designed heuristic approaches may actually be more acceptable to regulators than complex but opaque analytical methods. The key is ensuring that heuristics are:
Transparent: Decision logic should be clearly documented and explainable
Consistent: Similar situations should be handled similarly
Defensible: The rationale for the heuristic approach should be based on evidence and experience
Adaptive: The approach should evolve based on feedback and changing conditions
The Integration Challenge
The adaptive toolbox shouldn’t replace all analytical methods—it should complement them within a broader risk management framework. The key is understanding when to use which approach.
Use Heuristics When:
Time pressure is significant
Information is incomplete and unlikely to improve quickly
The decision context is familiar and patterns are recognizable
The consequences of being approximately right quickly outweigh being precisely right slowly
Resource constraints limit the feasibility of comprehensive analysis
Use Analytical Methods When:
Stakes are extremely high and errors could have catastrophic consequences
Time permits thorough analysis
The decision context is novel and patterns are unclear
Multiple stakeholders need to understand and agree on decision logic
Looking Forward
Gigerenzer’s work suggests that effective quality risk management will increasingly look like a hybrid approach that combines the best of analytical rigor with the adaptive flexibility of heuristic decision-making.
This evolution is already happening informally as quality professionals develop intuitive responses to common situations and use analytical methods primarily for novel or high-stakes decisions. The challenge is making this hybrid approach explicit and systematic rather than leaving it to individual discretion.
Future quality management systems will likely feature:
Adaptive Decision Support: Systems that learn from historical decisions and suggest appropriate heuristics for new situations
Context-Sensitive Approaches: Risk management methods that automatically adjust based on situational factors
Rapid Iteration Capabilities: Systems designed for quick adjustment rather than comprehensive upfront planning
Integrated Uncertainty Management: Approaches that explicitly acknowledge and work with uncertainty rather than trying to eliminate it
The Cultural Transformation
Perhaps the most significant challenge in implementing Gigerenzer’s insights isn’t technical—it’s cultural. Quality organizations have invested decades in building analytical capabilities and may resist approaches that appear to diminish the value of that investment.
The key to successful cultural transformation is demonstrating that heuristic approaches don’t eliminate analysis—they optimize it by focusing analytical effort where it provides the most value. This requires leadership that understands both the power and limitations of different decision-making approaches.
Organizations that successfully implement adaptive toolbox principles often find that they can:
Make decisions faster without sacrificing quality
Reduce analysis paralysis in routine situations
Free up analytical resources for genuinely complex problems
Improve decision consistency across teams
Adapt more quickly to changing conditions
Conclusion: Embracing Bounded Rationality
Gigerenzer’s adaptive toolbox offers a path forward that embraces rather than fights the reality of human cognition. By recognizing that our brains have evolved sophisticated mechanisms for making good decisions under uncertainty, we can develop quality systems that work with rather than against our cognitive strengths.
This doesn’t mean abandoning analytical rigor—it means applying it more strategically. It means recognizing that sometimes the best decision is the one made quickly with limited information rather than the one made slowly with comprehensive analysis. It means building systems that are robust to uncertainty rather than brittle in the face of incomplete information.
Most importantly, it means acknowledging that quality professionals are not computers. They are sophisticated pattern-recognition systems that have evolved to navigate uncertainty effectively. Our quality systems should amplify rather than override these capabilities.
The adaptive toolbox isn’t just a set of decision-making tools—it’s a different way of thinking about human rationality in organizational settings. For quality professionals willing to embrace this perspective, it offers the possibility of making better decisions, faster, with less stress and more confidence.
And in an industry where patient safety depends on the quality of our decisions, that possibility is worth pursuing, one heuristic at a time.
The pharmaceutical industry has long operated under what Michael Hudson aptly describes in his recent Forbes article as “symphonic control, “carefully orchestrated strategies executed with rigid precision, where quality units can function like conductors trying to control every note. But as Hudson observes, when our meticulously crafted risk assessments collide with chaotic reality, what emerges is often discordant. The time has come for quality risk management to embrace what I am going to call “rhythmic excellence,” a jazz-inspired approach that maintains rigorous standards while enabling adaptive performance in our increasingly BANI (Brittle, Anxious, Non-linear, and Incomprehensible) regulatory and manufacturing environment.
And since I love a good metaphor, I bring you:
Rhythmic Quality Risk Management
Recent research by Amy Edmondson and colleagues at Harvard Business School provides compelling evidence for rhythmic approaches to complex work. After studying more than 160 innovation teams, they found that performance suffered when teams mixed reflective activities (like risk assessments and control strategy development) with exploratory activities (like hazard identification and opportunity analysis) in the same time period. The highest-performing teams established rhythms that alternated between exploration and reflection, creating distinct beats for different quality activities.
This finding resonates deeply with the challenges we face in pharmaceutical quality risk management. Too often, our risk assessment meetings become frantic affairs where hazard identification, risk analysis, control strategy development, and regulatory communication all happen simultaneously. Teams push through these sessions exhausted and unsatisfied, delivering risk assessments they aren’t proud of—what Hudson describes as “cognitive whiplash”.
From Symphonic Control to Jazz-Based Quality Leadership
The traditional approach to pharmaceutical quality risk management mirrors what Hudson calls symphonic leadership—attempting to impose top-down structure as if more constraint and direction are what teams need to work with confidence. We create detailed risk assessment procedures, prescriptive FMEA templates, and rigid review schedules, then wonder why our teams struggle to adapt when new hazards emerge or when manufacturing conditions change unexpectedly.
Karl Weick’s work on organizational sensemaking reveals why this approach undermines our quality objectives: complex manufacturing environments require “mindful organizing” and the ability to notice subtle changes and respond fluidly. Setting a quality rhythm and letting go of excessive control provides support without constraint, giving teams the freedom to explore emerging risks, experiment with novel control strategies, and make sense of the quality challenges they face.
This represents a fundamental shift in how we conceptualize quality risk management leadership. Instead of being the conductor trying to orchestrate every risk assessment note, quality leaders should function as the rhythm section—establishing predictable beats that keep everyone synchronized while allowing individual expertise to flourish.
The Quality Rhythm Framework: Four Essential Beats
Drawing from Hudson’s research-backed insights and integrating them with ICH Q9(R1) requirements, I envision a Quality Rhythm Framework built on four essential beats:
Beat 1: Find Your Risk Cadence
Establish predictable rhythms that create temporal anchors for your quality team while maintaining ICH Q9 compliance. Weekly hazard identification sessions, daily deviation assessments, monthly control strategy reviews, and quarterly risk communication cycles aren’t just meetings—they’re the beats that keep everyone synchronized while allowing individual risk management expression.
The ICH Q9(R1) revision’s emphasis on proportional formality aligns perfectly with this rhythmic approach. High-risk processes require more frequent beats, while lower-risk areas can operate with extended rhythms. The key is consistency within each risk category, creating what Weick calls “structured flexibility”—the ability to respond creatively within clear boundaries.
Consider implementing these quality-specific rhythmic structures:
Daily Risk Pulse: Brief stand-ups focused on emerging quality signals—not comprehensive risk assessments, but awareness-building sessions that keep the team attuned to the manufacturing environment.
Weekly Hazard Identification Sessions: Dedicated time for exploring “what could go wrong” and, following ISO 31000 principles, “what could go better than expected.” These sessions should alternate between different product lines or process areas to maintain focus.
Monthly Control Strategy Reviews: Deeper evaluations of existing risk controls, including assessment of whether they remain appropriate and identification of optimization opportunities.
Quarterly Risk Communication Cycles: Structured information sharing with stakeholders, including regulatory bodies when appropriate, ensuring that risk insights flow effectively throughout the organization.
Beat 2: Pause for Quality Breaths
Hudson emphasizes that jazz musicians know silence is as important as sound, and quality risk management desperately needs structured pauses. Build quality breaths into your organizational rhythm—moments for reflection, integration, and recovery from the intense focus required for effective risk assessment.
Research by performance expert Jim Loehr demonstrates that sustainable excellence requires oscillation, not relentless execution. In quality contexts, this means creating space between intensive risk assessment activities and implementation of control strategies. These pauses allow teams to process complex risk information, integrate diverse perspectives, and avoid the decision fatigue that leads to poor risk judgments.
Practical quality breaths include:
Post-Assessment Integration Time: Following comprehensive risk assessments, build in periods where team members can reflect on findings, consult additional resources, and refine their thinking before finalizing control strategies.
Cross-Functional Synthesis Sessions: Regular meetings where different functions (Quality, Operations, Regulatory, Technical) come together not to make decisions, but to share perspectives and build collective understanding of quality risks.
Knowledge Capture Moments: Structured time for documenting lessons learned, updating risk models based on new experience, and creating institutional memory that enhances future risk assessments.
Beat 3: Encourage Quality Experimentation
Within your rhythmic structure, create psychological safety and confidence that team members can explore novel risk identification approaches without fear of hitting “wrong notes.” When learning and reflection are part of a predictable beat, trust grows and experimentation becomes part of the quality flow.
The ICH Q9(R1) revision’s focus on managing subjectivity in risk assessments creates opportunities for experimental approaches. Instead of viewing subjectivity as a problem to eliminate, we can experiment with structured methods for harnessing diverse perspectives while maintaining analytical rigor.
Hudson’s research shows that predictable rhythm facilitates innovation—when people are comfortable with the rhythm, they’re free to experiment with the melody. In quality risk management, this means establishing consistent frameworks that enable creative hazard identification and innovative control strategy development.
Experimental approaches might include:
Success Mode and Benefits Analysis (SMBA): As I’ve discussed previously, complement traditional FMEA with systematic identification of positive potential outcomes. Experiment with different SMBA formats and approaches to find what works best for specific process areas.
Cross-Industry Risk Insights: Dedicate portions of risk assessment sessions to exploring how other industries handle similar quality challenges. These experiments in perspective-taking can reveal blind spots in traditional pharmaceutical approaches.
Scenario-Based Risk Planning: Experiment with “what if” exercises that go beyond traditional failure modes to explore complex, interdependent risk situations that might emerge in dynamic manufacturing environments.
Beat 4: Enable Quality Solos
Just as jazz musicians trade solos while the ensemble provides support, look for opportunities for individual quality team members to drive specific risk management initiatives. This distributed leadership approach builds capability while maintaining collective coherence around quality objectives.
Hudson’s framework emphasizes that adaptive leaders don’t try to be conductors but create conditions for others to lead. In quality risk management, this means identifying team members with specific expertise or interest areas and empowering them to lead risk assessments in those domains.
Quality leadership solos might include:
Process Expert Risk Leadership: Assign experienced operators or engineers to lead risk assessments for processes they know intimately, with quality professionals providing methodological support.
Cross-Functional Risk Coordination: Empower individuals to coordinate risk management across organizational boundaries, taking ownership for ensuring all relevant perspectives are incorporated.
Innovation Risk Championship: Designate team members to lead risk assessments for new technologies or novel approaches, building expertise in emerging quality challenges.
The Rhythmic Advantage: Three Quality Transformation Benefits
Mastering these rhythmic approaches to quality risk management provide three advantages that mirror Hudson’s leadership research:
Fluid Quality Structure
A jazz ensemble can improvise because musicians share a rhythm. Similarly, quality rhythms keep teams functioning together while offering freedom to adapt to emerging risks, changing regulatory requirements, or novel manufacturing challenges. Management researchers call this “structured flexibility”—exactly what ICH Q9(R1) envisions when it emphasizes proportional formality.
When quality teams operate with shared rhythms, they can respond more effectively to unexpected events. A contamination incident doesn’t require completely reinventing risk assessment approaches—teams can accelerate their established rhythms, bringing familiar frameworks to bear on novel challenges while maintaining analytical rigor.
Sustainable Quality Energy
Quality risk management is inherently demanding work that requires sustained attention to complex, interconnected risks. Traditional approaches often lead to burnout as teams struggle with relentless pressure to identify every possible hazard and implement perfect controls. Rhythmic approaches prevent this exhaustion by regulating pace and integrating recovery.
More importantly, rhythmic quality management aligns teams around purpose and vision rather than merely compliance deadlines. This enables what performance researchers call “sustainable high performance”—quality excellence that endures rather than depletes organizational energy.
When quality professionals find rhythm in their risk management work, they develop what Mihaly Csikszentmihalyi identified as “flow state,” moments when attention is fully focused and performance feels effortless. These states are crucial for the deep thinking required for effective hazard identification and the creative problem-solving needed for innovative control strategies.
Enhanced Quality Trust and Innovation
The paradox Hudson identifies, that some constraint enables creativity, applies directly to quality risk management. Predictable rhythms don’t stifle innovation; they provide the stable foundation from which teams can explore novel approaches to quality challenges.
When quality teams know they have regular, structured opportunities for risk exploration, they’re more willing to raise difficult questions, challenge assumptions, and propose unconventional solutions. The rhythm creates psychological safety for intellectual risk-taking within the controlled environment of systematic risk assessment.
This enhanced innovation capability is particularly crucial as pharmaceutical manufacturing becomes increasingly complex, with continuous manufacturing, advanced process controls, and novel drug modalities creating quality challenges that traditional risk management approaches weren’t designed to address.
Integrating Rhythmic Principles with ICH Q9(R1) Compliance
The beauty of rhythmic quality risk management lies in its fundamental compatibility with ICH Q9(R1) requirements. The revision’s emphasis on scientific knowledge, proportional formality, and risk-based decision-making aligns perfectly with rhythmic approaches that create structured flexibility for quality teams.
Rhythmic Risk Assessment Enhancement
ICH Q9 requires systematic hazard identification, risk analysis, and risk evaluation. Rhythmic approaches enhance these activities by establishing regular, focused sessions for each component rather than trying to accomplish everything in marathon meetings.
During dedicated hazard identification beats, teams can employ diverse techniques—traditional brainstorming, structured what-if analysis, cross-industry benchmarking, and the Success Mode and Benefits Analysis I’ve advocated. The rhythm ensures these activities receive appropriate attention while preventing the cognitive overload that reduces identification effectiveness.
Risk analysis benefits from rhythmic separation between data gathering and interpretation activities. Teams can establish rhythms for collecting process data, manufacturing experience, and regulatory intelligence, followed by separate beats for analyzing this information and developing risk models.
Rhythmic Risk Control Development
The ICH Q9(R1) emphasis on risk-based decision-making aligns perfectly with rhythmic approaches to control strategy development. Instead of rushing from risk assessment to control implementation, rhythmic approaches create space for thoughtful strategy development that considers multiple options and their implications.
Rhythmic control development might include beats for:
Control Strategy Ideation: Creative sessions focused on generating potential control approaches without immediate evaluation of feasibility or cost.
Implementation Planning: Separate sessions for detailed planning of selected control strategies, including resource requirements, timeline development, and change management considerations.
Effectiveness Assessment: Regular rhythms for evaluating implemented controls, gathering performance data, and identifying optimization opportunities.
Rhythmic Risk Communication
ICH Q9’s communication requirements benefit significantly from rhythmic approaches. Instead of ad hoc communication when problems arise, establish regular rhythms for sharing risk insights, control strategy updates, and lessons learned.
Quality communication rhythms should align with organizational decision-making cycles, ensuring that risk insights reach stakeholders when they’re most useful for decision-making. This might include monthly updates to senior leadership, quarterly reports to regulatory affairs, and annual comprehensive risk reviews for long-term strategic planning.
Practical Implementation: Building Your Quality Rhythm
Implementing rhythmic quality risk management requires systematic integration rather than wholesale replacement of existing approaches. Start by evaluating your current risk management processes to identify natural rhythm points and opportunities for enhancement.
Phase 1: Rhythm Assessment and Planning
Map your existing quality risk management activities against rhythmic principles. Identify where teams experience the cognitive whiplash Hudson describes—trying to accomplish too many different types of thinking in single sessions. Look for opportunities to separate exploration from analysis, strategy development from implementation planning, and individual reflection from group decision-making.
Establish criteria for quality rhythm frequency based on risk significance, process complexity, and organizational capacity. High-risk processes might require daily pulse checks and weekly deep dives, while lower-risk areas might operate effectively with monthly assessment rhythms.
Train quality teams on rhythmic principles and their application to risk management. Help them understand how rhythm enhances rather than constrains their analytical capabilities, providing structure that enables deeper thinking and more creative problem-solving.
Phase 2: Pilot Program Development
Select pilot areas where rhythmic approaches are most likely to demonstrate clear benefits. New product development projects, technology implementation initiatives, or process improvement activities often provide ideal testing grounds because their inherent uncertainty creates natural opportunities for both risk management and opportunity identification.
Design pilot programs to test specific rhythmic principles:
Rhythm Separation: Compare traditional comprehensive risk assessment meetings with rhythmic approaches that separate hazard identification, risk analysis, and control strategy development into distinct sessions.
Quality Breathing: Experiment with structured pauses between intensive risk assessment activities and measure their impact on decision quality and team satisfaction.
Distributed Leadership: Identify opportunities for team members to lead specific aspects of risk management and evaluate the impact on engagement and expertise development.
Phase 3: Organizational Integration
Based on pilot results, develop systematic approaches for scaling rhythmic quality risk management across the organization. This requires integration with existing quality systems, regulatory processes, and organizational governance structures.
Consider how rhythmic approaches will interact with regulatory inspection activities, change control processes, and continuous improvement initiatives. Ensure that rhythmic flexibility doesn’t compromise documentation requirements or audit trail integrity.
Establish metrics for evaluating rhythmic quality risk management effectiveness, including both traditional risk management indicators (incident rates, control effectiveness, regulatory compliance) and rhythm-specific measures (team engagement, innovation frequency, decision speed).
Phase 4: Continuous Enhancement and Cultural Integration
Like all aspects of quality risk management, rhythmic approaches require continuous improvement based on experience and changing needs. Regular assessment of rhythm effectiveness helps refine approaches over time and ensures sustained benefits.
The ultimate goal is cultural integration—making rhythmic thinking a natural part of how quality professionals approach risk management challenges. This requires consistent leadership modeling, recognition of rhythmic successes, and integration of rhythmic principles into performance expectations and career development.
Measuring Rhythmic Quality Success
Traditional quality metrics focus primarily on negative outcome prevention: deviation rates, batch failures, regulatory findings, and compliance scores. While these remain important, rhythmic quality risk management requires expanded measurement approaches that capture both defensive effectiveness and adaptive capability.
Enhanced metrics should include:
Rhythm Consistency Indicators: Frequency of established quality rhythms, participation rates in rhythmic activities, and adherence to planned cadences.
Innovation and Adaptation Measures: Number of novel risk identification approaches tested, implementation rate of creative control strategies, and frequency of process improvements emerging from risk management activities.
Team Engagement and Development: Participation in quality leadership opportunities, cross-functional collaboration frequency, and professional development within risk management capabilities.
Decision Quality Indicators: Time from risk identification to control implementation, stakeholder satisfaction with risk communication, and long-term effectiveness of implemented controls.
Regulatory Considerations: Communicating Rhythmic Value
Regulatory agencies are increasingly interested in risk-based approaches that demonstrate genuine process understanding and continuous improvement capabilities. Rhythmic quality risk management strengthens regulatory relationships by showing sophisticated thinking about process optimization and quality enhancement within established frameworks.
When communicating with regulatory agencies, emphasize how rhythmic approaches improve process understanding, enhance control strategy development, and support continuous improvement objectives. Show how structured flexibility leads to better patient protection through more responsive and adaptive quality systems.
Focus regulatory communications on how enhanced risk understanding leads to better quality outcomes rather than on operational efficiency benefits that might appear secondary to regulatory objectives. Demonstrate how rhythmic approaches maintain analytical rigor while enabling more effective responses to emerging quality challenges.
The Future of Quality Risk Management: Beyond Rhythm to Resonance
As we master rhythmic approaches to quality risk management, the next evolution involves what I call “quality resonance”—the phenomenon that occurs when individual quality rhythms align and amplify each other across organizational boundaries. Just as musical instruments can create resonance that produces sounds more powerful than any individual instrument, quality organizations can achieve resonant states where risk management effectiveness transcends the sum of individual contributions.
Resonant quality organizations share several characteristics:
Synchronized Rhythm Networks: Quality rhythms in different departments, processes, and product lines align to create organization-wide patterns of risk awareness and response capability.
Harmonic Risk Communication: Information flows between quality functions create harmonics that amplify important signals while filtering noise, enabling more effective decision-making at all organizational levels.
Emergent Quality Intelligence: The interaction of multiple rhythmic quality processes generates insights and capabilities that wouldn’t be possible through individual efforts alone.
Building toward quality resonance requires sustained commitment to rhythmic principles, continuous refinement of quality cadences, and patient development of organizational capability. The payoff, however, is transformational: quality risk management that not only prevents problems but actively creates value through enhanced understanding, improved processes, and strengthened competitive position.
Finding Your Quality Beat
Uncertainty is inevitable in pharmaceutical manufacturing, regulatory environments, and global supply chains. As Hudson emphasizes, the choice is whether to exhaust ourselves trying to conduct every quality note or to lay down rhythms that enable entire teams to create something extraordinary together.
Tomorrow morning, when you walk into that risk assessment meeting, you’ll face this choice in real time. Will you pick up the conductor’s baton, trying to control every analytical voice? Or will you sit at the back of the stage and create the beat on which your quality team can find its flow?
The research is clear: rhythmic approaches to complex work create better outcomes, higher engagement, and more sustainable performance. The ICH Q9(R1) framework provides the flexibility needed to implement rhythmic quality risk management while maintaining regulatory compliance. The tools and techniques exist to transform quality risk management from a defensive necessity into an adaptive capability that drives innovation and competitive advantage.
The question isn’t whether rhythmic quality risk management will emerge—it’s whether your organization will lead this transformation or struggle to catch up. The teams that master quality rhythm first will be best positioned to thrive in our increasingly BANI pharmaceutical world, turning uncertainty into opportunity while maintaining the rigorous standards our patients deserve.
Start with one beat. Find one aspect of your current quality risk management where you can separate exploration from analysis, create space for reflection, or enable someone to lead. Feel the difference that rhythm makes. Then gradually expand, building the quality jazz ensemble that our complex manufacturing world demands.
The rhythm section is waiting. It’s time to find your quality beat.
The pharmaceutical industry has long operated under a fundamental epistemological fallacy that undermines our ability to truly understand the effectiveness of our quality systems. We celebrate zero deviations, zero recalls, zero adverse events, and zero regulatory observations as evidence that our systems are working. But a fundamental fact we tend to ignore is that we are confusing the absence of evidence with evidence of absence—a logical error that not only fails to prove effectiveness but actively impedes our ability to build more robust, science-based quality systems.
This challenge strikes at the heart of how we approach quality risk management. When our primary evidence of “success” is that nothing bad happened, we create unfalsifiable systems that can never truly be proven wrong.
The Philosophical Foundation: Falsifiability in Quality Risk Management
Karl Popper’s theory of falsification fundamentally challenges how we think about scientific validity. For Popper, the distinguishing characteristic of genuine scientific theories is not that they can be proven true, but that they can be proven false. A theory that cannot conceivably be refuted by any possible observation is not scientific—it’s metaphysical speculation.
Applied to quality risk management, this creates an uncomfortable truth: most of our current approaches to demonstrating system effectiveness are fundamentally unscientific. When we design quality systems around preventing negative outcomes and then use the absence of those outcomes as evidence of effectiveness, we create what Popper would call unfalsifiable propositions. No possible observation could ever prove our system ineffective as long as we frame effectiveness in terms of what didn’t happen.
Consider the typical pharmaceutical quality narrative: “Our manufacturing process is validated because we haven’t had any quality failures in twelve months.” This statement is unfalsifiable because it can always accommodate new information. If a failure occurs next month, we simply adjust our understanding of the system’s reliability without questioning the fundamental assumption that absence of failure equals validation. We might implement corrective actions, but we rarely question whether our original validation approach was capable of detecting the problems that eventually manifested.
Most of our current risk models are either highly predictive but untestable (making them useful for operational decisions but scientifically questionable) or neither predictive nor testable (making them primarily compliance exercises). The goal should be to move toward models are both scientifically rigorous and practically useful.
This philosophical foundation has practical implications for how we design and evaluate quality risk management systems. Instead of asking “How can we prevent bad things from happening?” we should be asking “How can we design systems that will fail in predictable ways when our underlying assumptions are wrong?” The first question leads to unfalsifiable defensive strategies; the second leads to falsifiable, scientifically valid approaches to quality assurance.
Why “Nothing Bad Happened” Isn’t Evidence of Effectiveness
The fundamental problem with using negative evidence to prove positive claims extends far beyond philosophical niceties, it creates systemic blindness that prevents us from understanding what actually drives quality outcomes. When we frame effectiveness in terms of absence, we lose the ability to distinguish between systems that work for the right reasons and systems that appear to work due to luck, external factors, or measurement limitations.
Scenario
Null Hypothesis
What Rejection Proves
What Non-Rejection Proves
Popperian Assessment
Traditional Efficacy Testing
No difference between treatment and control
Treatment is effective
Cannot prove effectiveness
Falsifiable and useful
Traditional Safety Testing
No increased risk
Treatment increases risk
Cannot prove safety
Unfalsifiable for safety
Absence of Events (Current)
No safety signal detected
Cannot prove anything
Cannot prove safety
Unfalsifiable
Non-inferiority Approach
Excess risk > acceptable margin
Treatment is acceptably safe
Cannot prove safety
Partially falsifiable
Falsification-Based Safety
Safety controls are inadequate
Current safety measures fail
Safety controls are adequate
Falsifiable and actionable
The table above demonstrates how traditional safety and effectiveness assessments fall into unfalsifiable categories. Traditional safety testing, for example, attempts to prove that something doesn’t increase risk, but this can never be definitively demonstrated—we can only fail to detect increased risk within the limitations of our study design. This creates a false confidence that may not be justified by the actual evidence.
The Sampling Illusion: When we observe zero deviations in a batch of 1000 units, we often conclude that our process is in control. But this conclusion conflates statistical power with actual system performance. With typical sampling strategies, we might have only 10% power to detect a 1% defect rate. The “zero observations” reflect our measurement limitations, not process capability.
The Survivorship Bias: Systems that appear effective may be surviving not because they’re well-designed, but because they haven’t yet encountered the conditions that would reveal their weaknesses. Our quality systems are often validated under ideal conditions and then extrapolated to real-world operations where different failure modes may dominate.
The Attribution Problem: When nothing bad happens, we attribute success to our quality systems without considering alternative explanations. Market forces, supplier improvements, regulatory changes, or simple random variation might be the actual drivers of observed outcomes.
Observable Outcome
Traditional Interpretation
Popperian Critique
What We Actually Know
Testable Alternative
Zero adverse events in 1000 patients
“The drug is safe”
Absence of evidence does not equal Evidence of absence
No events detected in this sample
Test limits of safety margin
Zero manufacturing deviations in 12 months
“The process is in control”
No failures observed does not equal a Failure-proof system
No deviations detected with current methods
Challenge process with stress conditions
Zero regulatory observations
“The system is compliant”
No findings does not equal No problems exist
No issues found during inspection
Audit against specific failure modes
Zero product recalls
“Quality is assured”
No recalls does not equal No quality issues
No quality failures reached market
Test recall procedures and detection
Zero patient complaints
“Customer satisfaction achieved”
No complaints does not equal No problems
No complaints received through channels
Actively solicit feedback mechanisms
This table illustrates how traditional interpretations of “positive” outcomes (nothing bad happened) fail to provide actionable knowledge. The Popperian critique reveals that these observations tell us far less than we typically assume, and the testable alternatives provide pathways toward more rigorous evaluation of system effectiveness.
The pharmaceutical industry’s reliance on these unfalsifiable approaches creates several downstream problems. First, it prevents genuine learning and improvement because we can’t distinguish effective interventions from ineffective ones. Second, it encourages defensive mindsets that prioritize risk avoidance over value creation. Third, it undermines our ability to make resource allocation decisions based on actual evidence of what works.
The Model Usefulness Problem: When Predictions Don’t Match Reality
George Box’s famous aphorism that “all models are wrong, but some are useful” provides a pragmatic framework for this challenge, but it doesn’t resolve the deeper question of how to determine when a model has crossed from “useful” to “misleading.” Popper’s falsifiability criterion offers one approach: useful models should make specific, testable predictions that could potentially be proven wrong by future observations.
The challenge in pharmaceutical quality management is that our models often serve multiple purposes that may be in tension with each other. Models used for regulatory submission need to demonstrate conservative estimates of risk to ensure patient safety. Models used for operational decision-making need to provide actionable insights for process optimization. Models used for resource allocation need to enable comparison of risks across different areas of the business.
When the same model serves all these purposes, it often fails to serve any of them well. Regulatory models become so conservative that they provide little guidance for actual operations. Operational models become so complex that they’re difficult to validate or falsify. Resource allocation models become so simplified that they obscure important differences in risk characteristics.
The solution isn’t to abandon modeling, but to be more explicit about the purpose each model serves and the criteria by which its usefulness should be judged. For regulatory purposes, conservative models that err on the side of safety may be appropriate even if they systematically overestimate risks. For operational decision-making, models should be judged primarily on their ability to correctly rank-order interventions by their impact on relevant outcomes. For scientific understanding, models should be designed to make falsifiable predictions that can be tested through controlled experiments or systematic observation.
Consider the example of cleaning validation, where we use models to predict the probability of cross-contamination between manufacturing campaigns. Traditional approaches focus on demonstrating that residual contamination levels are below acceptance criteria—essentially proving a negative. But this approach tells us nothing about the relative importance of different cleaning parameters, the margin of safety in our current procedures, or the conditions under which our cleaning might fail.
A more falsifiable approach would make specific predictions about how changes in cleaning parameters affect contamination levels. We might hypothesize that doubling the rinse time reduces contamination by 50%, or that certain product sequences create systematically higher contamination risks. These hypotheses can be tested and potentially falsified, providing genuine learning about the underlying system behavior.
From Defensive to Testable Risk Management
The evolution from defensive to testable risk management represents a fundamental shift in how we conceptualize quality systems. Traditional defensive approaches ask, “How can we prevent failures?” Testable approaches ask, “How can we design systems that fail predictably when our assumptions are wrong?” This shift moves us from unfalsifiable defensive strategies toward scientifically rigorous quality management.
This transition aligns with the broader evolution in risk thinking documented in ICH Q9(R1) and ISO 31000, which recognize risk as “the effect of uncertainty on objectives” where that effect can be positive, negative, or both. By expanding our definition of risk to include opportunities as well as threats, we create space for falsifiable hypotheses about system performance.
The integration of opportunity-based thinking with Popperian falsifiability creates powerful synergies. When we hypothesize that a particular quality intervention will not only reduce defects but also improve efficiency, we create multiple testable predictions. If the intervention reduces defects but doesn’t improve efficiency, we learn something important about the underlying system mechanics. If it improves efficiency but doesn’t reduce defects, we gain different insights. If it does neither, we discover that our fundamental understanding of the system may be flawed.
This approach requires a cultural shift from celebrating the absence of problems to celebrating the presence of learning. Organizations that embrace falsifiable quality management actively seek conditions that would reveal the limitations of their current systems. They design experiments to test the boundaries of their process capabilities. They view unexpected results not as failures to be explained away, but as opportunities to refine their understanding of system behavior.
The practical implementation of testable risk management involves several key elements:
Hypothesis-Driven Validation: Instead of demonstrating that processes meet specifications, validation activities should test specific hypotheses about process behavior. For example, rather than proving that a sterilization cycle achieves a 6-log reduction, we might test the hypothesis that cycle modifications affect sterility assurance in predictable ways. Instead of demonstrating that the CHO cell culture process consistently produces mAb drug substance meeting predetermined specifications, hypothesis-driven validation would test the specific prediction that maintaining pH at 7.0 ± 0.05 during the production phase will result in final titers that are 15% ± 5% higher than pH maintained at 6.9 ± 0.05, creating a falsifiable hypothesis that can be definitively proven wrong if the predicted titer improvement fails to materialize within the specified confidence intervals
Falsifiable Control Strategies: Control strategies should include specific predictions about how the system will behave under different conditions. These predictions should be testable and potentially falsifiable through routine monitoring or designed experiments.
Learning-Oriented Metrics: Key indicators should be designed to detect when our assumptions about system behavior are incorrect, not just when systems are performing within specification. Metrics that only measure compliance tell us nothing about the underlying system dynamics.
Proactive Stress Testing: Rather than waiting for problems to occur naturally, we should actively probe the boundaries of system performance through controlled stress conditions. This approach reveals failure modes before they impact patients while providing valuable data about system robustness.
Designing Falsifiable Quality Systems
The practical challenge of designing falsifiable quality systems requires a fundamental reconceptualization of how we approach quality assurance. Instead of building systems designed to prevent all possible failures, we need systems designed to fail in instructive ways when our underlying assumptions are incorrect.
This approach starts with making our assumptions explicit and testable. Traditional quality systems often embed numerous unstated assumptions about process behavior, material characteristics, environmental conditions, and human performance. These assumptions are rarely articulated clearly enough to be tested, making the systems inherently unfalsifiable. A falsifiable quality system makes these assumptions explicit and designs tests to evaluate their validity.
Consider the design of a typical pharmaceutical manufacturing process. Traditional approaches focus on demonstrating that the process consistently produces product meeting specifications under defined conditions. This demonstration typically involves process validation studies that show the process works under idealized conditions, followed by ongoing monitoring to detect deviations from expected performance.
A falsifiable approach would start by articulating specific hypotheses about what drives process performance. We might hypothesize that product quality is primarily determined by three critical process parameters, that these parameters interact in predictable ways, and that environmental variations within specified ranges don’t significantly impact these relationships. Each of these hypotheses can be tested and potentially falsified through designed experiments or systematic observation of process performance.
The key insight is that falsifiable quality systems are designed around testable theories of what makes quality systems effective, rather than around defensive strategies for preventing all possible problems. This shift enables genuine learning and continuous improvement because we can distinguish between interventions that work for the right reasons and those that appear to work for unknown or incorrect reasons.
Structured Hypothesis Formation: Quality requirements should be built around explicit hypotheses about cause-and-effect relationships in critical processes. These hypotheses should be specific enough to be tested and potentially falsified through systematic observation or experimentation.
Predictive Monitoring: Instead of monitoring for compliance with specifications, systems should monitor for deviations from predicted behavior. When predictions prove incorrect, this provides valuable information about the accuracy of our underlying process understanding.
Experimental Integration: Routine operations should be designed to provide ongoing tests of system hypotheses. Process changes, material variations, and environmental fluctuations should be treated as natural experiments that provide data about system behavior rather than disturbances to be minimized.
Failure Mode Anticipation: Quality systems should explicitly anticipate the ways failures might happen and design detection mechanisms for these failure modes. This proactive approach contrasts with reactive systems that only detect problems after they occur.
The Evolution of Risk Assessment: From Compliance to Science
The evolution of pharmaceutical risk assessment from compliance-focused activities to genuine scientific inquiry represents one of the most significant opportunities for improving quality outcomes. Traditional risk assessments often function primarily as documentation exercises designed to satisfy regulatory requirements rather than tools for genuine learning and improvement.
ICH Q9(R1) recognizes this limitation and calls for more scientifically rigorous approaches to quality risk management. The updated guidance emphasizes the need for risk assessments to be based on scientific knowledge and to provide actionable insights for quality improvement. This represents a shift away from checklist-based compliance activities toward hypothesis-driven scientific inquiry.
The integration of falsifiability principles with ICH Q9(R1) requirements creates opportunities for more rigorous and useful risk assessments. Instead of asking generic questions about what could go wrong, falsifiable risk assessments develop specific hypotheses about failure modes and design tests to evaluate these hypotheses. This approach provides more actionable insights while meeting regulatory expectations for systematic risk evaluation.
Consider the evolution of Failure Mode and Effects Analysis (FMEA) from a traditional compliance tool to a falsifiable risk assessment method. Traditional FMEA often devolves into generic lists of potential failures with subjective probability and impact assessments. The results provide limited insight because the assessments can’t be systematically tested or validated.
A falsifiable FMEA would start with specific hypotheses about failure mechanisms and their relationships to process parameters, material characteristics, or operational conditions. These hypotheses would be tested through historical data analysis, designed experiments, or systematic monitoring programs. The results would provide genuine insights into system behavior while creating a foundation for continuous improvement.
This evolution requires changes in how we approach several key risk assessment activities:
Hazard Identification: Instead of brainstorming all possible things that could go wrong, risk identification should focus on developing testable hypotheses about specific failure mechanisms and their triggers.
Risk Analysis: Probability and impact assessments should be based on testable models of system behavior rather than subjective expert judgment. When models prove inaccurate, this provides valuable information about the need to revise our understanding of system dynamics.
Risk Control: Control measures should be designed around testable theories of how interventions affect system behavior. The effectiveness of controls should be evaluated through systematic monitoring and periodic testing rather than assumed based on their implementation.
Risk Review: Risk review activities should focus on testing the accuracy of previous risk predictions and updating risk models based on new evidence. This creates a learning loop that continuously improves the quality of risk assessments over time.
Practical Framework for Falsifiable Quality Risk Management
The implementation of falsifiable quality risk management requires a systematic framework that integrates Popperian principles with practical pharmaceutical quality requirements. This framework must be sophisticated enough to generate genuine scientific insights while remaining practical for routine quality management activities.
The foundation of this framework rests on the principle that effective quality systems are built around testable theories of what drives quality outcomes. These theories should make specific predictions that can be evaluated through systematic observation, controlled experimentation, or historical data analysis. When predictions prove incorrect, this provides valuable information about the need to revise our understanding of system behavior.
Phase 1: Hypothesis Development
The first phase involves developing specific, testable hypotheses about system behavior. These hypotheses should address fundamental questions about what drives quality outcomes in specific operational contexts. Rather than generic statements about quality risks, hypotheses should make specific predictions about relationships between process parameters, material characteristics, environmental conditions, and quality outcomes.
For example, instead of the generic hypothesis that “temperature variations affect product quality,” a falsifiable hypothesis might state that “temperature excursions above 25°C for more than 30 minutes during the mixing phase increase the probability of out-of-specification results by at least 20%.” This hypothesis is specific enough to be tested and potentially falsified through systematic data collection and analysis.
Phase 2: Experimental Design
The second phase involves designing systematic approaches to test the hypotheses developed in Phase 1. This might involve controlled experiments, systematic analysis of historical data, or structured monitoring programs designed to capture relevant data about hypothesis validity.
The key principle is that testing approaches should be capable of falsifying the hypotheses if they are incorrect. This requires careful attention to statistical power, measurement systems, and potential confounding factors that might obscure true relationships between variables.
Phase 3: Evidence Collection
The third phase focuses on systematic collection of evidence relevant to hypothesis testing. This evidence might come from designed experiments, routine monitoring data, or systematic analysis of historical performance. The critical requirement is that evidence collection should be structured around hypothesis testing rather than generic performance monitoring.
Evidence collection systems should be designed to detect when hypotheses are incorrect, not just when systems are performing within specifications. This requires more sophisticated approaches to data analysis and interpretation than traditional compliance-focused monitoring.
Phase 4: Hypothesis Evaluation
The fourth phase involves systematic evaluation of evidence against the hypotheses developed in Phase 1. This evaluation should follow rigorous statistical methods and should be designed to reach definitive conclusions about hypothesis validity whenever possible.
When hypotheses are falsified, this provides valuable information about the need to revise our understanding of system behavior. When hypotheses are supported by evidence, this provides confidence in our current understanding while suggesting areas for further testing and refinement.
Phase 5: System Adaptation
The final phase involves adapting quality systems based on the insights gained through hypothesis testing. This might involve modifying control strategies, updating risk assessments, or redesigning monitoring programs based on improved understanding of system behavior.
The critical principle is that system adaptations should be based on genuine learning about system behavior rather than reactive responses to compliance issues or external pressures. This creates a foundation for continuous improvement that builds cumulative knowledge about what drives quality outcomes.
Implementation Challenges
The transition to falsifiable quality risk management faces several practical challenges that must be addressed for successful implementation. These challenges range from technical issues related to experimental design and statistical analysis to cultural and organizational barriers that may resist more scientifically rigorous approaches to quality management.
Technical Challenges
The most immediate technical challenge involves designing falsifiable hypotheses that are relevant to pharmaceutical quality management. Many quality professionals have extensive experience with compliance-focused activities but limited experience with experimental design and hypothesis testing. This skills gap must be addressed through targeted training and development programs.
Statistical power represents another significant technical challenge. Many quality systems operate with very low baseline failure rates, making it difficult to design experiments with adequate power to detect meaningful differences in system performance. This requires sophisticated approaches to experimental design and may necessitate longer observation periods or larger sample sizes than traditionally used in quality management.
Measurement systems present additional challenges. Many pharmaceutical quality attributes are difficult to measure precisely, introducing uncertainty that can obscure true relationships between process parameters and quality outcomes. This requires careful attention to measurement system validation and uncertainty quantification.
Cultural and Organizational Challenges
Perhaps more challenging than technical issues are the cultural and organizational barriers to implementing more scientifically rigorous quality management approaches. Many pharmaceutical organizations have deeply embedded cultures that prioritize risk avoidance and compliance over learning and improvement.
The shift to falsifiable quality management requires cultural change that embraces controlled failure as a learning opportunity rather than something to be avoided at all costs. This represents a fundamental change in how many organizations think about quality management and may encounter significant resistance.
Regulatory relationships present additional organizational challenges. Many quality professionals worry that more rigorous scientific approaches to quality management might raise regulatory concerns or create compliance burdens. This requires careful communication with regulatory agencies to demonstrate that falsifiable approaches enhance rather than compromise patient safety.
Strategic Solutions
Successfully implementing falsifiable quality risk management requires strategic approaches that address both technical and cultural challenges. These solutions must be tailored to specific organizational contexts while maintaining scientific rigor and regulatory compliance.
Pilot Programs: Implementation should begin with carefully selected pilot programs in areas where falsifiable approaches can demonstrate clear value. These pilots should be designed to generate success stories that support broader organizational adoption while building internal capability and confidence.
Training and Development: Comprehensive training programs should be developed to build organizational capability in experimental design, statistical analysis, and hypothesis testing. These programs should be tailored to pharmaceutical quality contexts and should emphasize practical applications rather than theoretical concepts.
Regulatory Engagement: Proactive engagement with regulatory agencies should emphasize how falsifiable approaches enhance patient safety through improved understanding of system behavior. This communication should focus on the scientific rigor of the approach rather than on business benefits that might appear secondary to regulatory objectives.
Cultural Change Management: Systematic change management programs should address cultural barriers to embracing controlled failure as a learning opportunity. These programs should emphasize how falsifiable approaches support regulatory compliance and patient safety rather than replacing these priorities with business objectives.
Case Studies: Falsifiability in Practice
The practical application of falsifiable quality risk management can be illustrated through several case studies that demonstrate how Popperian principles can be integrated with routine pharmaceutical quality activities. These examples show how hypotheses can be developed, tested, and used to improve quality outcomes while maintaining regulatory compliance.
Case Study 1: Cleaning Validation Optimization
A biologics manufacturer was experiencing occasional cross-contamination events despite having validated cleaning procedures that consistently met acceptance criteria. Traditional approaches focused on demonstrating that cleaning procedures reduced contamination below specified limits, but provided no insight into the factors that occasionally caused this system to fail.
The falsifiable approach began with developing specific hypotheses about cleaning effectiveness. The team hypothesized that cleaning effectiveness was primarily determined by three factors: contact time with cleaning solution, mechanical action intensity, and rinse water temperature. They further hypothesized that these factors interacted in predictable ways and that current procedures provided a specific margin of safety above minimum requirements.
These hypotheses were tested through a designed experiment that systematically varied each cleaning parameter while measuring residual contamination levels. The results revealed that current procedures were adequate under ideal conditions but provided minimal margin of safety when multiple factors were simultaneously at their worst-case levels within specified ranges.
Based on these findings, the cleaning procedure was modified to provide greater margin of safety during worst-case conditions. More importantly, ongoing monitoring was redesigned to test the continued validity of the hypotheses about cleaning effectiveness rather than simply verifying compliance with acceptance criteria.
Case Study 2: Process Control Strategy Development
A pharmaceutical manufacturer was developing a control strategy for a new manufacturing process. Traditional approaches would have focused on identifying critical process parameters and establishing control limits based on process validation studies. Instead, the team used a falsifiable approach that started with explicit hypotheses about process behavior.
The team hypothesized that product quality was primarily controlled by the interaction between temperature and pH during the reaction phase, that these parameters had linear effects on product quality within the normal operating range, and that environmental factors had negligible impact on these relationships.
These hypotheses were tested through systematic experimentation during process development. The results confirmed the importance of the temperature-pH interaction but revealed nonlinear effects that weren’t captured in the original hypotheses. More importantly, environmental humidity was found to have significant effects on process behavior under certain conditions.
The control strategy was designed around the revised understanding of process behavior gained through hypothesis testing. Ongoing process monitoring was structured to continue testing key assumptions about process behavior rather than simply detecting deviations from target conditions.
Case Study 3: Supplier Quality Management
A biotechnology company was managing quality risks from a critical raw material supplier. Traditional approaches focused on incoming inspection and supplier auditing to verify compliance with specifications and quality system requirements. However, occasional quality issues suggested that these approaches weren’t capturing all relevant quality risks.
The falsifiable approach started with specific hypotheses about what drove supplier quality performance. The team hypothesized that supplier quality was primarily determined by their process control during critical manufacturing steps, that certain environmental conditions increased the probability of quality issues, and that supplier quality system maturity was predictive of long-term quality performance.
These hypotheses were tested through systematic analysis of supplier quality data, enhanced supplier auditing focused on specific process control elements, and structured data collection about environmental conditions during material manufacturing. The results revealed that traditional quality system assessments were poor predictors of actual quality performance, but that specific process control practices were strongly predictive of quality outcomes.
The supplier management program was redesigned around the insights gained through hypothesis testing. Instead of generic quality system requirements, the program focused on specific process control elements that were demonstrated to drive quality outcomes. Supplier performance monitoring was structured around testing continued validity of the relationships between process control and quality outcomes.
Measuring Success in Falsifiable Quality Systems
The evaluation of falsifiable quality systems requires fundamentally different approaches to performance measurement than traditional compliance-focused systems. Instead of measuring the absence of problems, we need to measure the presence of learning and the accuracy of our predictions about system behavior.
Traditional quality metrics focus on outcomes: defect rates, deviation frequencies, audit findings, and regulatory observations. While these metrics remain important for regulatory compliance and business performance, they provide limited insight into whether our quality systems are actually effective or merely lucky. Falsifiable quality systems require additional metrics that evaluate the scientific validity of our approach to quality management.
Predictive Accuracy Metrics
The most direct measure of a falsifiable quality system’s effectiveness is the accuracy of its predictions about system behavior. These metrics evaluate how well our hypotheses about quality system behavior match observed outcomes. High predictive accuracy suggests that we understand the underlying drivers of quality outcomes. Low predictive accuracy indicates that our understanding needs refinement.
Predictive accuracy metrics might include the percentage of process control predictions that prove correct, the accuracy of risk assessments in predicting actual quality issues, or the correlation between predicted and observed responses to process changes. These metrics provide direct feedback about the validity of our theoretical understanding of quality systems.
Learning Rate Metrics
Another important category of metrics evaluates how quickly our understanding of quality systems improves over time. These metrics measure the rate at which falsified hypotheses lead to improved system performance or more accurate predictions. High learning rates indicate that the organization is effectively using falsifiable approaches to improve quality outcomes.
Learning rate metrics might include the time required to identify and correct false assumptions about system behavior, the frequency of successful process improvements based on hypothesis testing, or the rate of improvement in predictive accuracy over time. These metrics evaluate the dynamic effectiveness of falsifiable quality management approaches.
Hypothesis Quality Metrics
The quality of hypotheses generated by quality risk management processes represents another important performance dimension. High-quality hypotheses are specific, testable, and relevant to important quality outcomes. Poor-quality hypotheses are vague, untestable, or focused on trivial aspects of system performance.
Hypothesis quality can be evaluated through structured peer review processes, assessment of testability and specificity, and evaluation of relevance to critical quality attributes. Organizations with high-quality hypothesis generation processes are more likely to gain meaningful insights from their quality risk management activities.
System Robustness Metrics
Falsifiable quality systems should become more robust over time as learning accumulates and system understanding improves. Robustness can be measured through the system’s ability to maintain performance despite variations in operating conditions, changes in materials or equipment, or other sources of uncertainty.
Robustness metrics might include the stability of process performance across different operating conditions, the effectiveness of control strategies under stress conditions, or the system’s ability to detect and respond to emerging quality risks. These metrics evaluate whether falsifiable approaches actually lead to more reliable quality systems.
Regulatory Implications and Opportunities
The integration of falsifiable principles with pharmaceutical quality risk management creates both challenges and opportunities in regulatory relationships. While some regulatory agencies may initially view scientific approaches to quality management with skepticism, the ultimate result should be enhanced regulatory confidence in quality systems that can demonstrate genuine understanding of what drives quality outcomes.
The key to successful regulatory engagement lies in emphasizing how falsifiable approaches enhance patient safety rather than replacing regulatory compliance with business optimization. Regulatory agencies are primarily concerned with patient safety and product quality. Falsifiable quality systems support these objectives by providing more rigorous and reliable approaches to ensuring quality outcomes.
Enhanced Regulatory Submissions
Regulatory submissions based on falsifiable quality systems can provide more compelling evidence of system effectiveness than traditional compliance-focused approaches. Instead of demonstrating that systems meet minimum requirements, falsifiable approaches can show genuine understanding of what drives quality outcomes and how systems will behave under different conditions.
This enhanced evidence can support regulatory flexibility in areas such as process validation, change control, and ongoing monitoring requirements. Regulatory agencies may be willing to accept risk-based approaches to these activities when they’re supported by rigorous scientific evidence rather than generic compliance activities.
Proactive Risk Communication
Falsifiable quality systems enable more proactive and meaningful communication with regulatory agencies about quality risks and mitigation strategies. Instead of reactive communication about compliance issues, organizations can engage in scientific discussions about system behavior and improvement strategies.
This proactive communication can build regulatory confidence in organizational quality management capabilities while providing opportunities for regulatory agencies to provide input on scientific approaches to quality improvement. The result should be more collaborative regulatory relationships based on shared commitment to scientific rigor and patient safety.
Regulatory Science Advancement
The pharmaceutical industry’s adoption of more scientifically rigorous approaches to quality management can contribute to the advancement of regulatory science more broadly. Regulatory agencies benefit from industry innovations in risk assessment, process understanding, and quality assurance methods.
Organizations that successfully implement falsifiable quality risk management can serve as case studies for regulatory guidance development and can provide evidence for the effectiveness of science-based approaches to quality assurance. This contribution to regulatory science advancement creates value that extends beyond individual organizational benefits.
Toward a More Scientific Quality Culture
The long-term vision for falsifiable quality risk management extends beyond individual organizational implementations to encompass fundamental changes in how the pharmaceutical industry approaches quality assurance. This vision includes more rigorous scientific approaches to quality management, enhanced collaboration between industry and regulatory agencies, and continuous advancement in our understanding of what drives quality outcomes.
Industry-Wide Learning Networks
One promising direction involves the development of industry-wide learning networks that share insights from falsifiable quality management implementations. These networks facilitate collaborative hypothesis testing, shared learning from experimental results, and development of common methodologies for scientific approaches to quality assurance.
Such networks accelerate the advancement of quality science while maintaining appropriate competitive boundaries. Organizations should share methodological insights and general findings while protecting proprietary information about specific processes or products. The result would be faster advancement in quality management science that benefits the entire industry.
Advanced Analytics Integration
The integration of advanced analytics and machine learning techniques with falsifiable quality management approaches represents another promising direction. These technologies can enhance our ability to develop testable hypotheses, design efficient experiments, and analyze complex datasets to evaluate hypothesis validity.
Machine learning approaches are particularly valuable for identifying patterns in complex quality datasets that might not be apparent through traditional analysis methods. However, these approaches must be integrated with falsifiable frameworks to ensure that insights can be validated and that predictive models can be systematically tested and improved.
Regulatory Harmonization
The global harmonization of regulatory approaches to science-based quality management represents a significant opportunity for advancing patient safety and regulatory efficiency. As individual regulatory agencies gain experience with falsifiable quality management approaches, there are opportunities to develop harmonized guidance that supports consistent global implementation.
ICH Q9(r1) was a great step. I would love to see continued work in this area.
Embracing the Discomfort of Scientific Rigor
The transition from compliance-focused to scientifically rigorous quality risk management represents more than a methodological change—it requires fundamentally rethinking how we approach quality assurance in pharmaceutical manufacturing. By embracing Popper’s challenge that genuine scientific theories must be falsifiable, we move beyond the comfortable but ultimately unhelpful world of proving negatives toward the more demanding but ultimately more rewarding world of testing positive claims about system behavior.
The effectiveness paradox that motivates this discussion—the problem of determining what works when our primary evidence is that “nothing bad happened”—cannot be resolved through better compliance strategies or more sophisticated documentation. It requires genuine scientific inquiry into the mechanisms that drive quality outcomes. This inquiry must be built around testable hypotheses that can be proven wrong, not around defensive strategies that can always accommodate any possible outcome.
The practical implementation of falsifiable quality risk management is not without challenges. It requires new skills, different cultural approaches, and more sophisticated methodologies than traditional compliance-focused activities. However, the potential benefits—genuine learning about system behavior, more reliable quality outcomes, and enhanced regulatory confidence—justify the investment required for successful implementation.
Perhaps most importantly, the shift to falsifiable quality management moves us toward a more honest assessment of what we actually know about quality systems versus what we merely assume or hope to be true. This honesty is uncomfortable but essential for building quality systems that genuinely serve patient safety rather than organizational comfort.
The question is not whether pharmaceutical quality management will eventually embrace more scientific approaches—the pressures of regulatory evolution, competitive dynamics, and patient safety demands make this inevitable. The question is whether individual organizations will lead this transition or be forced to follow. Those that embrace the discomfort of scientific rigor now will be better positioned to thrive in a future where quality management is evaluated based on genuine effectiveness rather than compliance theater.
As we continue to navigate an increasingly complex regulatory and competitive environment, the organizations that master the art of turning uncertainty into testable knowledge will be best positioned to deliver consistent quality outcomes while maintaining the flexibility needed for innovation and continuous improvement. The integration of Popperian falsifiability with modern quality risk management provides a roadmap for achieving this mastery while maintaining the rigorous standards our industry demands.
The path forward requires courage to question our current assumptions, discipline to design rigorous tests of our theories, and wisdom to learn from both our successes and our failures. But for those willing to embrace these challenges, the reward is quality systems that are not only compliant but genuinely effective. Systems that we can defend not because they’ve never been proven wrong, but because they’ve been proven right through systematic, scientific inquiry.
The pharmaceutical industry has long operated under a defensive mindset when it comes to risk management. We identify what could go wrong, assess the likelihood and impact of failure modes, and implement controls to prevent or mitigate negative outcomes. This approach, while necessary and required by ICH Q9, represents only half the risk equation. What our quality risk management program could become not just a compliance necessity, but a strategic driver of innovation, efficiency, and competitive advantage?
Enter the ISO 31000 perspective on risk—one that recognizes risk as “the effect of uncertainty on objectives,” where that effect can be positive, negative, or both. This broader definition opens up transformative possibilities for how we approach quality risk management in pharmaceutical manufacturing. Rather than solely focusing on preventing bad things from happening, we can start identifying and capitalizing on good things that might occur.
The Evolution of Risk Thinking in Pharmaceuticals
For decades, our industry’s risk management approach has been shaped by regulatory necessity and liability concerns. The introduction of ICH Q9 in 2005—and its recent revision in 2023—provided a structured framework for quality risk management that emphasizes scientific knowledge, proportional formality, and patient protection. This framework has served us well, establishing systematic approaches to risk assessment, control, communication, and review.
However, the updated ICH Q9(R1) recognizes that we’ve been operating with significant blind spots. The revision addresses issues including “high levels of subjectivity in risk assessments,” “failing to adequately manage supply and product availability risks,” and “lack of clarity on risk-based decision-making”. These challenges suggest that our traditional approach to risk management, while compliant, may not be fully leveraging the strategic value that comprehensive risk thinking can provide.
The ISO 31000 standard offers a complementary perspective that can address these gaps. By defining risk as uncertainty’s effect on objectives—with explicit recognition that this effect can create opportunities as well as threats—ISO 31000 provides a framework for risk management that is inherently more strategic and value-creating.
Understanding Risk as Opportunity in the Pharmaceutical Context
Lot us start by establishing a clear understanding of what “positive risk” or “opportunity” means in our context. In pharmaceutical quality management, opportunities are uncertain events or conditions that, if they occur, would enhance our ability to achieve quality objectives beyond our current expectations.
Consider these examples:
Manufacturing Process Opportunities: A new analytical method validates faster than anticipated, allowing for reduced testing cycles and increased throughput. The uncertainty around validation timelines created an opportunity that, when realized, improved operational efficiency while maintaining quality standards.
Supply Chain Opportunities: A raw material supplier implements process improvements that result in higher-purity ingredients at lower cost. This positive deviation from expected quality created opportunities for enhanced product stability and improved margins.
Technology Integration Opportunities: Implementation of process analytical technology (PAT) tools not only meets their intended monitoring purpose but reveals previously unknown process insights that enable further optimization opportunities.
Regulatory Opportunities: A comprehensive quality risk assessment submitted as part of a regulatory filing demonstrates such thorough understanding of the product and process that regulators grant additional manufacturing flexibility, creating opportunities for more efficient operations.
These scenarios illustrate how uncertainty—the foundation of all risk—can work in our favor when we’re prepared to recognize and capitalize on positive outcomes.
The Strategic Value of Opportunity-Based Risk Management
Integrating opportunity recognition into your quality risk management program delivers value across multiple dimensions:
Enhanced Innovation Capability
Traditional risk management often creates conservative cultures where “safe” decisions are preferred over potentially transformative ones. By systematically identifying and evaluating opportunities, we can make more balanced decisions that account for both downside risks and upside potential. This leads to greater willingness to explore innovative approaches to quality challenges while maintaining appropriate risk controls.
Improved Resource Allocation
When we only consider negative risks, we tend to over-invest in protective measures while under-investing in value-creating activities. Opportunity-oriented risk management helps optimize resource allocation by identifying where investments might yield unexpected benefits beyond their primary purpose.
Strengthened Competitive Position
Companies that effectively identify and capitalize on quality-related opportunities can develop competitive advantages through superior operational efficiency, faster time-to-market, enhanced product quality, or innovative approaches to regulatory compliance.
Cultural Transformation
Perhaps most importantly, embracing opportunities transforms the perception of risk management from a necessary burden to a strategic enabler. This cultural shift encourages proactive thinking, innovation, and continuous improvement throughout the organization.
Mapping ISO 31000 Principles to ICH Q9 Requirements
The beauty of integrating ISO 31000’s opportunity perspective with ICH Q9 compliance lies in their fundamental compatibility. Both frameworks emphasize systematic, science-based approaches to risk management with proportional formality based on risk significance. The key difference is scope—ISO 31000’s broader definition of risk naturally encompasses opportunities alongside threats.
Risk Assessment Enhancement
ICH Q9 requires risk assessment to include hazard identification, analysis, and evaluation. The ISO 31000 approach enhances this by expanding identification beyond failure modes to include potential positive outcomes. During hazard analysis and risk assessment (HARA), we can systematically ask not only “what could go wrong?” but also “what could go better than expected?” and “what positive outcomes might emerge from this uncertainty?”
For example, when assessing risks associated with implementing a new manufacturing technology, traditional ICH Q9 assessment would focus on potential failures, integration challenges, and validation risks. The enhanced approach would also identify opportunities for improved process understanding, unexpected efficiency gains, or novel approaches to quality control that might emerge during implementation.
Risk Control Expansion
ICH Q9’s risk control phase traditionally focuses on risk reduction and risk acceptance. The ISO 31000 perspective adds a third dimension: opportunity enhancement. This involves implementing controls or strategies that not only mitigate negative risks but also position the organization to capitalize on positive uncertainties should they occur.
Consider controls designed to manage analytical method transfer risks. Traditional controls might include extensive validation studies, parallel testing, and contingency procedures. Opportunity-enhanced controls might also include structured data collection protocols designed to identify process insights, cross-training programs that build broader organizational capabilities, or partnerships with equipment vendors that could lead to preferential access to new technologies.
Risk Communication and Opportunity Awareness
ICH Q9 emphasizes the importance of risk communication among stakeholders. When we expand this to include opportunity communication, we create organizational awareness of positive possibilities that might otherwise go unrecognized. This enhanced communication helps ensure that teams across the organization are positioned to identify and report positive deviations that could represent valuable opportunities.
Risk Review and Opportunity Capture
The risk review process required by ICH Q9 becomes more dynamic when it includes opportunity assessment. Regular reviews should evaluate not only whether risk controls remain effective, but also whether any positive outcomes have emerged that could be leveraged for further benefit. This creates a feedback loop that continuously enhances both risk management and opportunity realization.
Implementation Framework
Implementing opportunity-based risk management within your existing ICH Q9 program requires systematic integration rather than wholesale replacement. Here’s a practical framework for making this transition:
Phase 1: Assessment and Planning
Begin by evaluating your current risk management processes to identify integration points for opportunity assessment. Review existing risk assessments to identify cases where positive outcomes might have been overlooked. Establish criteria for what constitutes a meaningful opportunity in your context—this might include potential cost savings, quality improvements, efficiency gains, or innovation possibilities above defined thresholds.
Key activities include:
Mapping current risk management processes against ISO 31000 principles
Perform a readiness evaluation
Training risk management teams on opportunity identification techniques
Developing templates and tools that prompt opportunity consideration
Establishing metrics for tracking opportunity identification and realization
Readiness Evaluation
Before implementing opportunity-based risk management, conduct a thorough assessment of organizational readiness and capability. This includes evaluating current risk management maturity, cultural factors that might support or hinder adoption, and existing processes that could be enhanced.
Key assessment areas include:
Current risk management process effectiveness and consistency
Organizational culture regarding innovation and change
Leadership support for expanded risk management approaches
Available resources for training and process enhancement
Systematically integrate opportunity assessment into your existing risk management workflows. This doesn’t require new procedures—rather, it involves enhancing existing processes to ensure opportunity identification receives appropriate attention alongside threat assessment.
Modify risk assessment templates to include opportunity identification sections. Train teams to ask opportunity-focused questions during risk identification sessions. Develop criteria for evaluating opportunity significance using similar approaches to threat assessment—considering likelihood, impact, and detectability.
Update risk control strategies to include opportunity enhancement alongside risk mitigation. This might involve designing controls that serve dual purposes or implementing monitoring systems that can detect positive deviations as well as negative ones.
This is the phase I am currently working through. Make sure to do a pilot program!
Pilot Program Development
Start with pilot programs in areas where opportunities are most likely to be identified and realized. This might include new product development projects, technology implementation initiatives, or process improvement activities where uncertainty naturally creates both risks and opportunities.
Design pilot programs to:
Test opportunity identification and evaluation methods
Develop organizational capability and confidence
Create success stories that support broader adoption
Refine processes and tools based on practical experience
Phase 3: Cultural Integration
The success of opportunity-based risk management ultimately depends on cultural adoption. Teams need to feel comfortable identifying and discussing positive possibilities without being perceived as overly optimistic or insufficiently rigorous.
Establish communication protocols that encourage opportunity reporting alongside issue escalation. Recognize and celebrate cases where teams successfully identify and capitalize on opportunities. Incorporate opportunity realization into performance metrics and success stories.
Scaling and Integration Strategy
Based on pilot program results, develop a systematic approach for scaling opportunity-based risk management across the organization. This should include timelines, resource requirements, training programs, and change management strategies.
Consider factors such as:
Process complexity and risk management requirements in different areas
Organizational change capacity and competing priorities
Resource availability and investment requirements
Integration with other improvement and innovation initiatives
Phase 4: Continuous Enhancement
Like all aspects of quality risk management, opportunity integration requires continuous improvement. Regular assessment of the program’s effectiveness in identifying and capitalizing on opportunities helps refine the approach over time.
Conduct periodic reviews of opportunity identification accuracy—are teams successfully recognizing positive outcomes when they occur? Evaluate opportunity realization effectiveness—when opportunities are identified, how successfully does the organization capitalize on them? Use these insights to enhance training, processes, and organizational support for opportunity-based risk management.
Long-term Sustainability Planning
Ensure that opportunity-based risk management becomes embedded in organizational culture and processes rather than remaining dependent on individual champions or special programs. This requires systematic integration into standard operating procedures, performance metrics, and leadership expectations.
Plan for:
Ongoing training and capability development programs
Regular assessment and continuous improvement of opportunity identification processes
Integration with career development and advancement criteria
Long-term resource allocation and organizational support
Tools and Techniques for Opportunity Integration
Include a Success Mode and Benefits Analysis in your FMEA (Failure Mode and Effects Analysis)
Traditional FMEA focuses on potential failures and their effects. Opportunity-enhanced FMEA includes “Success Mode and Benefits Analysis” (SMBA) that systematically identifies potential positive outcomes and their benefits. For each process step, teams assess not only what could go wrong, but also what could go better than expected and how to position the organization to benefit from such outcomes.
A Success Mode and Benefits Analysis (SMBA) is the positive complement to the traditional Failure Mode and Effects Analysis (FMEA). While FMEA identifies where things can go wrong and how to prevent or mitigate failures, SMBA systematically evaluates how things can go unexpectedly right—helping organizations proactively capture, enhance, and realize benefits that arise from process successes, innovations, or positive deviations.
What Does a Success Mode and Benefits Analysis Look Like?
The SMBA is typically structured as a table or worksheet with a format paralleling the FMEA, but with a focus on positive outcomes and opportunities. A typical SMBA process includes the following columns and considerations:
Step/Column
Description
Process Step/Function
The specific process, activity, or function under investigation.
Success Mode
Description of what could go better than expected or intended—what’s the positive deviation?
Benefits/Effects
The potential beneficial effects if the success mode occurs (e.g., improved yield, faster cycle, enhanced quality, regulatory flexibility).
Likelihood (L)
Estimated probability that the success mode will occur.
Magnitude of Benefit (M)
Qualitative or quantitative evaluation of how significant the benefit would be (e.g., minor, moderate, major; or by quantifiable metrics).
Detectability
Can the opportunity be spotted early? What are the triggers or signals of this benefit occurring?
Actions to Capture/Enhance
Steps or controls that could help ensure the success is recognized and benefits are realized (e.g., monitoring plans, training, adaptation of procedures).
Benefit Priority Number (BPN)
An optional calculated field (e.g., L × M) to help the team prioritize follow-up actions.
Proactive Opportunity Identification: Instead of waiting for positive results to emerge, the process prompts teams to seek out “what could go better than planned?”.
Systematic Benefit Analysis: Quantifies or qualifies benefits just as FMEA quantifies risk.
Follow-Up Actions: Establishes ways to amplify and institutionalize successes.
When and How to Use SMBA
Use SMBA alongside FMEA during new technology introductions, process changes, or annual reviews.
Integrate into cross-functional risk assessments to balance risk aversion with innovation.
Use it to foster a culture that not just “prevents failure,” but actively “captures opportunity” and learns from success.
Opportunity-Integrated Risk Matrices
Traditional risk matrices plot likelihood versus impact for negative outcomes. Enhanced matrices include separate quadrants or scales for positive outcomes, allowing teams to visualize both threats and opportunities in the same framework. This provides a more complete picture of uncertainty and helps prioritize actions based on overall risk-opportunity balance.
Scenario Planning with Upside Cases
While scenario planning typically focuses on “what if” situations involving problems, opportunity-oriented scenario planning includes “what if” situations involving unexpected successes. This helps teams prepare to recognize and capitalize on positive outcomes that might otherwise be missed.
Innovation-Focused Risk Assessments
When evaluating new technologies, processes, or approaches, include systematic assessment of innovation opportunities that might emerge. This involves considering not just whether the primary objective will be achieved, but what secondary benefits or unexpected capabilities might develop during implementation.
Organizational Considerations
Leadership Commitment and Cultural Change
Successful integration of opportunity-based risk management requires genuine leadership commitment to cultural change. Leaders must model behavior that values both threat mitigation and opportunity creation. This means celebrating teams that identify valuable opportunities alongside those that prevent significant risks.
Leadership should establish clear expectations that risk management includes opportunity identification as a core responsibility. Performance metrics, recognition programs, and resource allocation decisions should reflect this balanced approach to uncertainty management.
Training and Capability Development
Teams need specific training to develop opportunity identification skills. While threat identification often comes naturally in quality-conscious cultures, opportunity recognition requires different cognitive approaches and tools.
Training programs should include:
Techniques for identifying positive potential outcomes
Methods for evaluating opportunity significance and likelihood
Approaches for designing controls that enhance opportunities while mitigating risks
Communication skills for discussing opportunities without compromising analytical rigor
Cross-Functional Integration
Opportunity-based risk management is most effective when integrated across organizational functions. Quality teams might identify process improvement opportunities, while commercial teams recognize market advantages, and technical teams discover innovation possibilities.
Establishing cross-functional opportunity review processes ensures that identified opportunities receive appropriate evaluation and resource allocation regardless of their origin. Regular communication between functions helps build organizational capability to recognize and act on opportunities systematically.
Measuring Success in Opportunity-Based Risk Management
Existing risk management metrics typically focus on negative outcome prevention: deviation rates, incident frequency, compliance scores, and similar measures. While these remain important, opportunity-based programs should also track positive outcome realization.
Enhanced metrics might include:
Number of opportunities identified per risk assessment
Percentage of identified opportunities that are successfully realized
Value generated from opportunity realization (cost savings, quality improvements, efficiency gains)
Time from opportunity identification to realization
Innovation and Improvement Indicators
Opportunity-focused risk management should drive increased innovation and continuous improvement. Tracking metrics related to process improvements, technology adoption, and innovation initiatives provides insight into the program’s effectiveness in creating value beyond compliance.
Consider monitoring:
Rate of process improvement implementation
Success rate of new technology adoptions
Number of best practices developed and shared across the organization
Frequency of positive deviations that lead to process optimization
Cultural and Behavioral Measures
The ultimate success of opportunity-based risk management depends on cultural integration. Measuring changes in organizational attitudes, behaviors, and capabilities provides insight into program sustainability and long-term impact.
Relevant measures include:
Employee engagement with risk management processes
Frequency of voluntary opportunity reporting
Cross-functional collaboration on risk and opportunity initiatives
Leadership participation in opportunity evaluation and resource allocation
Regulatory Considerations and Compliance Integration
Maintaining ICH Q9 Compliance
The opportunity-enhanced approach must maintain full compliance with ICH Q9 requirements while adding value through expanded scope. This means ensuring that all required elements of risk assessment, control, communication, and review continue to receive appropriate attention and documentation.
Regulatory submissions should clearly demonstrate that opportunity identification enhances rather than compromises systematic risk evaluation. Documentation should show how opportunity assessment strengthens process understanding and control strategy development.
Communicating Value to Regulators
Regulators are increasingly interested in risk-based approaches that demonstrate genuine process understanding and continuous improvement capabilities. Opportunity-based risk management can strengthen regulatory relationships by demonstrating sophisticated thinking about process optimization and quality enhancement.
When communicating with regulatory agencies, emphasize how opportunity identification improves process understanding, enhances control strategy development, and supports continuous improvement objectives. Show how the approach leads to better risk control through deeper process knowledge and more robust quality systems.
Global Harmonization Considerations
Different regulatory regions may have varying levels of comfort with opportunity-focused risk management discussions. While the underlying risk management activities remain consistent with global standards, communication approaches should be tailored to regional expectations and preferences.
Focus regulatory communications on how enhanced risk understanding leads to better patient protection and product quality, rather than on business benefits that might appear secondary to regulatory objectives.
Conclusion
Integrating ISO 31000’s opportunity perspective with ICH Q9 compliance represents more than a process enhancement and is a shift toward strategic risk management that positions quality organizations as value creators rather than cost centers. By systematically identifying and capitalizing on positive uncertainties, we can transform quality risk management from a defensive necessity into an offensive capability that drives innovation, efficiency, and competitive advantage.
The framework outlined here provides a practical path forward that maintains regulatory compliance while unlocking the strategic value inherent in comprehensive risk thinking. Success requires leadership commitment, cultural change, and systematic implementation, but the potential returns—in terms of operational excellence, innovation capability, and competitive position—justify the investment.
As we continue to navigate an increasingly complex and uncertain business environment, organizations that master the art of turning uncertainty into opportunity will be best positioned to thrive. The integration of ISO 31000’s risk-as-opportunities approach with ICH Q9 compliance provides a roadmap for achieving this mastery while maintaining the rigorous standards our industry demands.
The concept of “buying down risk” through operational capability development fundamentally depends on addressing the cognitive foundations that underpin effective risk assessment and decision-making. There are three critical systematic vulnerabilities that plague risk management processes: unjustified assumptions, incomplete identification of risks, and inappropriate use of risk assessment tools. These failures represent more than procedural deficiencies—they expose cognitive and knowledge management vulnerabilities that can undermine even the most well-intentioned quality systems.
Unjustified assumptions emerge when organizations rely on historical performance data or familiar process knowledge without adequately considering how changes in conditions, equipment, or supply chains might alter risk profiles. This manifests through anchoring bias, where teams place undue weight on initial information, leading to conclusions like “This process has worked safely for five years, so the risk profile remains unchanged.” Confirmation bias compounds this issue by causing assessors to seek information confirming existing beliefs while ignoring contradictory evidence.
Incomplete risk identification occurs when cognitive limitations and organizational biases inhibit comprehensive hazard recognition. Availability bias leads to overemphasis on dramatic but unlikely events while underestimating more probable but less memorable risks. Additionally, groupthink in risk assessment teams causes initial dissenting voices to be suppressed as consensus builds around preferred conclusions, limiting the scope of risks considered.
Inappropriate use of risk assessment tools represents the third systematic vulnerability, where organizations select methodologies based on familiarity rather than appropriateness for specific decision-making contexts. This includes using overly formal tools for trivial issues, applying generic assessment approaches without considering specific operational contexts, and relying on subjective risk scoring that provides false precision without meaningful insight. The misapplication often leads to risk assessments that fail to add value or clarity because they only superficially address root causes while generating high levels of subjectivity and uncertainty in outputs.
Traditional risk management approaches often focus on methodological sophistication while overlooking the cognitive realities that determine assessment effectiveness. Risk management operates fundamentally as a framework rather than a rigid methodology, providing structural architecture that enables systematic approaches to identifying, assessing, and controlling uncertainties. This framework distinction proves crucial because it recognizes that excellence emerges from the intersection of systematic process design with cognitive support systems that work with, rather than against, human decision-making patterns.
The Minimal Viable Risk Assessment Team: Beyond Compliance Theater
The foundation of cognitive excellence in risk management begins with assembling teams designed for cognitive rigor, knowledge depth, and psychological safety rather than mere compliance box-checking. The minimal viable risk assessment team concept challenges traditional approaches by focusing on four non-negotiable core roles that provide essential cognitive perspectives and knowledge anchors.
The Four Cognitive Anchors
Process Owner: The Reality Anchor represents lived operational experience rather than signature authority. This individual has engaged with the operation within the last 90 days and carries authority to change methods, budgets, and training. Authentic process ownership dismantles assumptions by grounding every risk statement in current operational facts, countering the tendency toward unjustified assumptions that plague many risk assessments.
Molecule Steward: The Patient’s Advocate moves beyond generic subject matter expertise to provide specific knowledge of how the particular product fails and can translate deviations into patient impact. When temperature drifts during freeze-drying, the molecule steward can explain whether a monoclonal antibody will aggregate or merely lose shelf life. Without this anchor, teams inevitably under-score hazards that never appear in generic assessment templates.
Technical System Owner: The Engineering Interpreter bridges the gap between equipment design intentions and operational realities. Equipment obeys physics rather than meeting minutes, and the system owner must articulate functional requirements, design limits, and engineering principles. This role prevents method-focused teams from missing systemic failures where engineering and design flaws could push entire batches outside critical parameters.
Quality Integrator: The Bias Disruptor forces cross-functional dialogue and preserves evidence of decision-making processes. Quality’s mission involves writing assumption logs, challenging confirmation bias, and ensuring dissenting voices are heard. This role maintains knowledge repositories so future teams are not condemned to repeat forgotten errors, directly addressing the knowledge management dimension of systematic risk assessment failure.
Knowledge Accessibility: The Missing Link in Risk Management
The Knowledge Accessibility Index (KAI) provides a systematic framework for evaluating how effectively organizations can access and deploy critical knowledge when decision-making requires specialized expertis. Unlike traditional knowledge management metrics focusing on knowledge creation or storage, the KAI specifically evaluates the availability, retrievability, and usability of knowledge at the point of decision-making.
Four Dimensions of Knowledge Accessibility
Expert Knowledge Availability assesses whether organizations can identify and access subject matter experts when specialized knowledge is required. This includes expert mapping and skill matrices, availability assessment during different operational scenarios, knowledge succession planning, and cross-training coverage for critical capabilities. The pharmaceutical environment demands that a qualified molecule steward be accessible within two hours for critical quality decisions, yet many organizations lack systematic approaches to ensuring this availability.
Knowledge Retrieval Efficiency measures how quickly and effectively teams can locate relevant information when making decisions. This encompasses search functionality effectiveness, knowledge organization and categorization, information architecture alignment with decision-making workflows, and access permissions balancing protection with accessibility. Time to find information represents a critical efficiency indicator that directly impacts the quality of risk assessment outcomes.
Knowledge Quality and Currency evaluates whether accessible knowledge is accurate, complete, and up-to-date through information accuracy verification processes, knowledge update frequency management, source credibility validation mechanisms, and completeness assessment relative to decision-making requirements. Outdated or incomplete knowledge can lead to systematic assessment failures even when expertise appears readily available.
Contextual Applicability assesses whether knowledge can be effectively applied to specific decision-making contexts through knowledge contextualization for operational scenarios, applicability assessment for different situations, integration capabilities with existing processes, and usability evaluation from end-user perspectives. Knowledge that exists but cannot be effectively applied provides little value during critical risk assessment activities.
Effective risk assessment team design fundamentally serves as knowledge preservation, not just compliance fulfillment. Every effective risk team is a living repository of organizational critical process insights, technical know-how, and operational experience. When teams include process owners, technical system engineers, molecule stewards, and quality integrators with deep hands-on familiarity, they collectively safeguard hard-won lessons and tacit knowledge that are often lost during organizational transitions.
Combating organizational forgetting requires intentional, cross-functional team design that fosters active knowledge transfer. When risk teams bring together diverse experts who routinely interact, challenge assumptions, and share context from respective domains, they create dynamic environments where critical information is surfaced, scrutinized, and retained. This living dialogue proves more effective than static records because it allows continuous updating and contextualization of knowledge in response to new challenges, regulatory changes, and operational shifts.
Team design becomes a strategic defense against the silent erosion of expertise that can leave organizations exposed to avoidable risks. By prioritizing teams that embody both breadth and depth of experience, organizations create robust safety nets that catch subtle warning signs, adapt to evolving risks, and ensure critical knowledge endures beyond individual tenure. This transforms collective memory into competitive advantage and foundation for sustained quality.
Cultural Integration: Embedding Cognitive Excellence
The development of truly effective risk management capabilities requires cultural transformation that embeds cognitive excellence principles into organizational DNA. Organizations with strong risk management cultures demonstrate superior capability in preventing quality issues, detecting problems early, and implementing effective corrective actions that address root causes rather than symptoms.
Psychological Safety as Cognitive Infrastructure
Psychological safety creates the foundational environment where personnel feel comfortable challenging assumptions, raising concerns about potential risks, and admitting uncertainty or knowledge limitations. This requires organizational cultures that treat questioning and systematic analysis as valuable contributions rather than obstacles to efficiency. Without psychological safety, the most sophisticated risk assessment methodologies and team compositions cannot overcome the fundamental barrier of information suppression.
Leaders must model vulnerability by sharing personal errors and how systems, not individuals, failed. They must invite dissent early in meetings with questions like “What might we be overlooking?” and reward candor by recognizing people who halt production over questionable trends. Psychological safety converts silent observers into active risk sensors, dramatically improving the effectiveness of knowledge accessibility and risk identification processes.
Structured Decision-Making as Cultural Practice
Excellence in pharmaceutical quality systems requires moving beyond hoping individuals will overcome cognitive limitations through awareness alone. Instead, organizations must design structured decision-making processes that systematically counter known biases while supporting comprehensive risk identification and analysis.
Forced systematic consideration involves checklists, templates, and protocols requiring teams to address specific risk categories and evidence types before reaching conclusions. Rather than relying on free-form discussion influenced by availability bias or groupthink, these tools ensure comprehensive coverage of relevant factors.
Devil’s advocate processes systematically introduce alternative perspectives and challenge preferred conclusions. By assigning specific individuals to argue against prevailing views or identify overlooked risks, organizations counter confirmation bias and overconfidence while identifying blind spots.
Staged decision-making separates risk identification from evaluation, preventing premature closure and ensuring adequate time for comprehensive hazard identification before moving to analysis and control decisions.
Implementation Framework: Building Cognitive Resilience
Phase 1: Knowledge Accessibility Audit
Organizations must begin with systematic knowledge accessibility audits that identify potential vulnerabilities in expertise availability and access. This audit addresses expertise mapping to identify knowledge holders and capabilities, knowledge accessibility assessment evaluating how effectively relevant knowledge can be accessed, knowledge quality evaluation assessing currency and completeness, and cognitive bias vulnerability assessment identifying situations where biases most likely affect conclusions.
For pharmaceutical manufacturing organizations, this audit might assess whether teams can access qualified molecule stewards within two hours for critical quality decisions, whether current system architecture documentation is accessible and comprehensible to risk assessment teams, whether process owners with recent operational experience are available for participation, and whether quality professionals can effectively challenge assumptions and integrate diverse perspectives.
Phase 2: Team Charter and Competence Framework
Moving from compliance theater to protection requires assembling teams with clear charters focused on cognitive rigor rather than checklist completion. An excellent risk team exists to frame, analyze, and communicate uncertainty so businesses can make science-based, patient-centered decisions. Before naming people, organizations must document the decisions teams must enable, the degree of formality those decisions demand, and the resources management will guarantee.
Competence proving rather than role filling ensures each core seat demonstrates documented capabilities. The process owner must have lived the operation recently with authority to change methods and budgets. The molecule steward must understand how specific products fail and translate deviations into patient impact. The technical system owner must articulate functional requirements and design limits. The quality integrator must force cross-functional dialogue and preserve evidence.
Phase 3: Knowledge System Integration
Knowledge-enabled decision making requires structures that make relevant information accessible at decision points while supporting cognitive processes necessary for accurate analysis. This involves structured knowledge capture that explicitly identifies assumptions, limitations, and context rather than simply documenting conclusions. Knowledge validation systems systematically test assumptions embedded in organizational knowledge, including processes for challenging accepted wisdom and updating mental models when new evidence emerges.
Expertise networks connect decision-makers with relevant specialized knowledge when required rather than relying on generalist teams for all assessments. Decision support systems prompt systematic consideration of potential biases and alternative explanations, creating technological infrastructure that supports rather than replaces human cognitive capabilities.
The final phase focuses on embedding cognitive excellence principles into organizational culture through systematic training programs that build both technical competencies and cognitive skills. These programs address not just what tools to use but how to think systematically about complex risk assessment challenges.
Continuous improvement mechanisms systematically analyze risk assessment performance to identify enhancement opportunities and implement improvements in methodologies, training, and support systems. Organizations track prediction accuracy, compare expected versus actual detectability, and feed insights into updated templates and training so subsequent teams start with enhanced capabilities.
Advanced Maturity: Predictive Risk Intelligence
Organizations achieving the highest levels of cognitive excellence implement predictive analytics, real-time bias detection, and adaptive systems that learn from assessment performance. These capabilities enable anticipation of potential risks and bias patterns before they manifest in assessment failures, including systematic monitoring of assessment performance, early warning systems for cognitive failures, and proactive adjustment of assessment approaches based on accumulated experience.
Adaptive learning systems continuously improve organizational capabilities based on performance feedback and changing conditions. These systems identify emerging patterns in risk assessment challenges and automatically adjust methodologies, training programs, and support systems to maintain effectiveness. Organizations at this maturity level contribute to industry knowledge and best practices while serving as benchmarks for other organizations.
From Reactive Compliance to Proactive Capability
The integration of cognitive science insights, knowledge accessibility frameworks, and team design principles creates a transformative approach to pharmaceutical risk management that moves beyond traditional compliance-focused activities toward strategic capability development. Organizations implementing these integrated approaches develop competitive advantages that extend far beyond regulatory compliance.
They build capabilities in systematic decision-making that improve performance across all aspects of pharmaceutical quality management. They create resilient systems that adapt to changing conditions while maintaining consistent effectiveness. Most importantly, they develop cultures of excellence that attract and retain exceptional talent while continuously improving capabilities.
The strategic integration of risk management practices with cultural transformation represents not merely an operational improvement opportunity but a fundamental requirement for sustained success in the evolving pharmaceutical manufacturing environment. Organizations implementing comprehensive risk buy-down strategies through systematic capability development will emerge as industry leaders capable of navigating regulatory complexity while delivering consistent value to patients, stakeholders, and society.
Excellence in this context means designing quality systems that work with human cognitive capabilities rather than against them. This requires integrating knowledge management principles with cognitive science insights to create environments where systematic, evidence-based decision-making becomes natural and sustainable. True elegance in quality system design comes from seamlessly integrating technical excellence with cognitive support, creating systems where the right decisions emerge naturally from the intersection of human expertise and systematic process.
Building Operational Capabilities Through Strategic Risk Management and Cultural Transformation
The Strategic Imperative: Beyond Compliance Theater
The fundamental shift from checklist-driven compliance to sustainable operational excellence grounded in robust risk management culture. Organizations continue to struggle with fundamental capability gaps that manifest as systemic compliance failures, operational disruptions, and ultimately, compromised patient safety.
The Risk Buy-Down Paradigm in Operations
The core challenge here is to build operational capabilities through proactively building systemic competencies that reduce the probability and impact of operational failures over time. Unlike traditional risk mitigation strategies that focus on reactive controls, risk buy-down emphasizes capability development that creates inherent resilience within operational systems.
This paradigm shifts the traditional cost-benefit equation from reactive compliance expenditure to proactive capability investment. Organizations implementing risk buy-down strategies recognize that upfront investments in operational excellence infrastructure generate compounding returns through reduced deviation rates, fewer regulatory observations, improved operational efficiency, and enhanced competitive positioning.
Economic Logic: Investment versus Failure Costs
The financial case for operational capability investment becomes stark when examining failure costs across the pharmaceutical industry. Drug development failures, inclusive of regulatory compliance issues, represent costs ranging from $500 to $900 million per program when accounting for capital costs and failure probabilities. Manufacturing quality failures trigger cascading costs including batch losses, investigation expenses, remediation efforts, regulatory responses, and market disruption.
Pharmaceutical manufacturers continue experiencing fundamental quality system failures despite decades of regulatory enforcement. These failures indicate insufficient investment in underlying operational capabilities, resulting in recurring compliance issues that generate exponentially higher long-term costs than proactive capability development would require.
Organizations successfully implementing risk buy-down strategies demonstrate measurable operational improvements. Companies with strong risk management cultures experience 30% higher likelihood of outperforming competitors while achieving 21% increases in productivity. These performance differentials reflect the compound benefits of systematic capability investment over reactive compliance expenditure.
Just look at the recent whitepaper published by the FDA to see the identified returns to this investment.
Regulatory Intelligence Framework Integration
The regulatory intelligence framework provides crucial foundation for risk buy-down implementation by enabling organizations to anticipate, assess, and proactively address emerging compliance requirements. Rather than responding reactively to regulatory observations, organizations with mature regulatory intelligence capabilities identify systemic capability gaps before they manifest as compliance violations.
Effective regulatory intelligence programs monitor FDA warning letter trends, 483 observations, and enforcement actions to identify patterns indicating capability deficiencies across industry segments. For example, persistent Quality Unit oversight failures across multiple geographic regions indicate fundamental organizational design issues rather than isolated procedural lapses8. This intelligence enables organizations to invest in Quality Unit empowerment, authority structures, and oversight capabilities before experiencing regulatory action.
The integration of regulatory intelligence with risk buy-down strategies creates a proactive capability development cycle where external regulatory trends inform internal capability investments, reducing both regulatory exposure and operational risk while enhancing competitive positioning through superior operational performance.
Culture as the Primary Risk Control
Organizational Culture as Foundational Risk Management
Organizational culture represents the most fundamental risk control mechanism within pharmaceutical operations, directly influencing how quality decisions are made, risks are identified and escalated, and operational excellence is sustained over time. Unlike procedural controls that can be circumvented or technical systems that can fail, culture operates as a pervasive influence that shapes behavior across all organizational levels and operational contexts.
Research demonstrates that organizations with strong risk management cultures are significantly less likely to experience damaging operational risk events and are better positioned to effectively respond when issues do occur.
The foundational nature of culture as a risk control becomes evident when examining quality system failures across pharmaceutical operations. Recent FDA warning letters consistently identify cultural deficiencies underlying technical violations, including insufficient Quality Unit authority, inadequate management commitment to compliance, and systemic failures in risk identification and escalation. These patterns indicate that technical compliance measures alone cannot substitute for robust quality culture.
Quality Culture Impact on Operational Resilience
Quality culture directly influences operational resilience by determining how organizations identify, assess, and respond to quality-related risks throughout manufacturing operations. Organizations with mature quality cultures demonstrate superior capability in preventing quality issues, detecting problems early, and implementing effective corrective actions that address root causes rather than symptoms.
Research in the biopharmaceutical industry reveals that integrating safety and quality cultures creates a unified “Resilience Culture” that significantly enhances organizational ability to sustain high-quality outcomes even under challenging conditions. This resilience culture is characterized by commitment to excellence, customer satisfaction focus, and long-term success orientation that transcends short-term operational pressures.
The operational impact of quality culture manifests through multiple mechanisms. Strong quality cultures promote proactive risk identification where employees at all levels actively surface potential quality concerns before they impact product quality. These cultures support effective escalation processes where quality issues receive appropriate priority regardless of operational pressures. Most importantly, mature quality cultures sustain continuous improvement mindsets where operational challenges become opportunities for systematic capability enhancement.
Dual-Approach Model: Leadership and Employee Ownership
Effective quality culture development requires coordinated implementation of top-down leadership commitment and bottom-up employee ownership, creating organizational alignment around quality principles and operational excellence. This dual-approach model recognizes that sustainable culture transformation cannot be achieved through leadership mandate alone, nor through grassroots initiatives without executive support.
Top-down leadership commitment establishes organizational vision, resource allocation, and accountability structures necessary for quality culture development. Research indicates that leadership commitment is vital for quality culture success and sustainability, with senior management responsible for initiating transformational change, setting quality vision, dedicating resources, communicating progress, and exhibiting visible support. Middle managers and supervisors ensure employees receive direct support and are held accountable to quality values.
Bottom-up employee ownership develops through empowerment, engagement, and competency development that enables staff to integrate quality considerations into daily operations. Organizations achieve employee ownership by incorporating quality into staff orientations, including quality expectations in job descriptions and performance appraisals, providing ongoing training opportunities, granting decision-making authority, and eliminating fear of consequences for quality-related concerns.
The integration of these approaches creates organizational conditions where quality culture becomes self-reinforcing. Leadership demonstrates commitment through resource allocation and decision-making priorities, while employees experience empowerment to make quality-focused decisions without fear of negative consequences for raising concerns or stopping production when quality issues arise.
Culture’s Role in Risk Identification and Response
Mature quality cultures fundamentally alter organizational approaches to risk identification and response by creating psychological safety for surfacing concerns, establishing systematic processes for risk assessment, and maintaining focus on long-term quality outcomes over short-term operational pressures. These cultural characteristics enable organizations to identify and address quality risks before they impact product quality or regulatory compliance.
Risk identification effectiveness depends critically on organizational culture that encourages transparency, values diverse perspectives, and rewards proactive concern identification. Research demonstrates that effective risk cultures promote “speaking up” where employees feel confident raising concerns and leaders demonstrate transparency in decision-making. This cultural foundation enables early risk detection that prevents minor issues from escalating into major quality failures.
Risk response effectiveness reflects cultural values around accountability, continuous improvement, and systematic problem-solving. Organizations with strong risk cultures implement thorough root cause analysis, develop comprehensive corrective and preventive actions, and monitor implementation effectiveness over time. These cultural practices ensure that risk responses address underlying causes rather than symptoms, preventing issue recurrence and building organizational learning capabilities.
The measurement of cultural risk management effectiveness requires systematic assessment of cultural indicators including employee engagement, incident reporting rates, management response to concerns, and the quality of corrective action implementation. Organizations tracking these cultural metrics can identify areas requiring improvement and monitor progress in cultural maturity over time.
Continuous Improvement Culture and Adaptive Capacity
Continuous improvement culture represents a fundamental organizational capability that enables sustained operational excellence through systematic enhancement of processes, systems, and capabilities over time. This culture creates adaptive capacity by embedding improvement mindsets, methodologies, and practices that enable organizations to evolve operational capabilities in response to changing requirements and emerging challenges.
Research demonstrates that continuous improvement culture significantly enhances operational performance through multiple mechanisms. Organizations with strong continuous improvement cultures experience increased employee engagement, higher productivity levels, enhanced innovation, and superior customer satisfaction. These performance improvements reflect the compound benefits of systematic capability development over time.
The development of continuous improvement culture requires systematic investment in employee competencies, improvement methodologies, data collection and analysis capabilities, and organizational learning systems. Organizations achieving mature improvement cultures provide training in improvement methodologies, establish improvement project pipelines, implement measurement systems that track improvement progress, and create recognition systems that reward improvement contributions.
Adaptive capacity emerges from continuous improvement culture through organizational learning mechanisms that capture knowledge from improvement projects, codify successful practices, and disseminate learning across the organization. This learning capability enables organizations to build institutional knowledge that improves response effectiveness to future challenges while preventing recurrence of past issues.
Integration with Regulatory Intelligence and Preventive Action
The integration of continuous improvement methodologies with regulatory intelligence capabilities creates proactive capability development systems that identify and address potential compliance issues before they manifest as regulatory observations. This integration represents advanced maturity in organizational quality management where external regulatory trends inform internal improvement priorities.
Regulatory intelligence provides continuous monitoring of FDA warning letters, 483 observations, enforcement actions, and guidance documents to identify emerging compliance trends and requirements. This intelligence enables organizations to anticipate regulatory expectations and proactively develop capabilities that address potential compliance gaps before they are identified through inspection.
Trending analysis of regulatory observations across industry segments reveals systemic capability gaps that multiple organizations experience. For example, persistent citations for Quality Unit oversight failures indicate industry-wide challenges in Quality Unit empowerment, authority structures, and oversight effectiveness. Organizations with mature regulatory intelligence capabilities use this trending data to assess their own Quality Unit capabilities and implement improvements before experiencing regulatory action.
The implementation of preventive action based on regulatory intelligence creates competitive advantage through superior regulatory preparedness while reducing compliance risk exposure. Organizations systematically analyzing regulatory trends and implementing capability improvements demonstrate regulatory readiness that supports inspection success and enables focus on operational excellence rather than compliance remediation.
The Integration Framework
Aligning Risk Management with Operational Capability Development
The strategic alignment of risk management principles with operational capability development creates synergistic organizational systems where risk identification enhances operational performance while operational excellence reduces risk exposure. This integration requires systematic design of management systems that embed risk considerations into operational processes while using operational data to inform risk management decisions.
Risk-based quality management approaches provide structured frameworks for integrating risk assessment with quality management processes throughout pharmaceutical operations. These approaches move beyond traditional compliance-focused quality management toward proactive systems that identify, assess, and mitigate quality risks before they impact product quality or regulatory compliance.
The implementation of risk-based approaches requires organizational capabilities in risk identification, assessment, prioritization, and mitigation that must be developed through systematic training, process development, and technology implementation. Organizations achieving mature risk-based quality management demonstrate superior performance in preventing quality issues, reducing deviation rates, and maintaining regulatory compliance.
Operational capability development supports risk management effectiveness by creating robust processes, competent personnel, and effective oversight systems that reduce the likelihood of risk occurrence while enhancing response effectiveness when risks do materialize. This capability development includes technical competencies, management systems, and organizational culture elements that collectively create operational resilience.
Efficiency-Excellence-Resilience Nexus
The strategic integration of efficiency, excellence, and resilience objectives creates organizational capabilities that simultaneously optimize resource utilization, maintain high-quality standards, and sustain performance under challenging conditions. This integration challenges traditional assumptions that efficiency and quality represent competing objectives, instead demonstrating that properly designed systems achieve superior performance across all dimensions.
Operational efficiency emerges from systematic elimination of waste, optimization of processes, and effective resource utilization that reduces operational costs while maintaining quality standards.
Operational excellence encompasses consistent achievement of high-quality outcomes through robust processes, competent personnel, and effective management systems.
Operational resilience represents the capability to maintain performance under stress, adapt to changing conditions, and recover effectively from disruptions. Resilience emerges from the integration of efficiency and excellence capabilities with adaptive capacity, redundancy planning, and organizational learning systems that enable sustained performance across varying conditions.
Measurement and Monitoring of Cultural Risk Management
The development of comprehensive measurement systems for cultural risk management enables organizations to track progress, identify improvement opportunities, and demonstrate the business value of culture investments. These measurement systems must capture both quantitative indicators of cultural effectiveness and qualitative assessments of cultural maturity across organizational levels.
Quantitative cultural risk management metrics include employee engagement scores, incident reporting rates, training completion rates, corrective action effectiveness measures, and regulatory compliance indicators. These metrics provide objective measures of cultural performance that can be tracked over time and benchmarked against industry standards.
Qualitative cultural assessment approaches include employee surveys, focus groups, management interviews, and observational assessments that capture cultural nuances not reflected in quantitative metrics. These qualitative approaches provide insights into cultural strengths, improvement opportunities, and the effectiveness of cultural transformation initiatives.
The integration of quantitative and qualitative measurement approaches creates comprehensive cultural assessment capabilities that inform management decision-making while demonstrating progress in cultural maturity. Organizations with mature cultural measurement systems can identify cultural risk indicators early, implement targeted interventions, and track improvement effectiveness over time.
Risk culture measurement frameworks must align with organizational risk appetite, regulatory requirements, and business objectives to ensure relevance and actionability. Effective frameworks establish clear definitions of desired cultural behaviors, implement systematic measurement processes, and create feedback mechanisms that inform continuous improvement in cultural effectiveness.
Common Capability Gaps Revealed Through FDA Observations
Analysis of FDA warning letters and 483 observations reveals persistent capability gaps across pharmaceutical manufacturing operations that reflect systemic weaknesses in organizational design, management systems, and quality culture. These capability gaps manifest as recurring regulatory observations that persist despite repeated enforcement actions, indicating fundamental deficiencies in operational capabilities rather than isolated procedural failures.
Quality Unit oversight failures represent the most frequently cited deficiency in FDA warning letters. These failures encompass insufficient authority to ensure CGMP compliance, inadequate resources for effective oversight, poor documentation practices, and systematic failures in deviation investigation and corrective action implementation. The persistence of Quality Unit deficiencies across multiple geographic regions indicates industry-wide challenges in Quality Unit design and empowerment.
Data integrity violations represent another systematic capability gap revealed through regulatory observations, including falsified records, inappropriate data manipulation, deleted electronic records, and inadequate controls over data generation and review. These violations indicate fundamental weaknesses in data governance systems, personnel training, and organizational culture around data integrity principles.
Deviation investigation and corrective action deficiencies appear consistently across FDA warning letters, reflecting inadequate capabilities in root cause analysis, corrective action development, and implementation effectiveness monitoring. These deficiencies indicate systematic weaknesses in problem-solving methodologies, investigation competencies, and management systems for tracking corrective action effectiveness.
Manufacturing process control deficiencies including inadequate validation, insufficient process monitoring, and poor change control implementation represent persistent capability gaps that directly impact product quality and regulatory compliance. These deficiencies reflect inadequate technical capabilities, insufficient management oversight, and poor integration between manufacturing and quality systems.
GMP Culture Translation to Operational Resilience
The five pillars of GMP – People, Product, Process, Procedures, and Premises – provide comprehensive framework for organizational capability development that addresses all aspects of pharmaceutical manufacturing operations. Effective GMP culture ensures that each pillar receives appropriate attention and investment while maintaining integration across all operational elements.
Personnel competency development represents the foundational element of GMP culture, encompassing technical training, quality awareness, regulatory knowledge, and continuous learning capabilities that enable employees to make appropriate quality decisions across varying operational conditions. Organizations with mature GMP cultures invest systematically in personnel development while creating career advancement opportunities that retain quality expertise.
Process robustness and validation ensure that manufacturing operations consistently produce products meeting quality specifications while providing confidence in process capability under normal operating conditions. GMP culture emphasizes process understanding, validation effectiveness, and continuous monitoring that enables proactive identification and resolution of process issues before they impact product quality.
Documentation systems and data integrity support all aspects of GMP implementation by providing objective evidence of compliance with regulatory requirements while enabling effective investigation and corrective action when issues occur. Mature GMP cultures emphasize documentation accuracy, completeness, and accessibility while implementing controls that prevent data integrity issues.
Risk-Based Quality Management as Operational Capability
Risk-based quality management represents advanced organizational capability that integrates risk assessment principles with quality management processes to create proactive systems that prevent quality issues while optimizing resource allocation. This capability enables organizations to focus quality oversight activities on areas with greatest potential impact while maintaining comprehensive quality assurance across all operations.
The implementation of risk-based quality management requires organizational capabilities in risk identification, assessment, prioritization, and mitigation that must be developed through systematic training, process development, and technology implementation. Organizations achieving mature risk-based capabilities demonstrate superior performance in preventing quality issues, reducing deviation rates, and maintaining regulatory compliance efficiency.
Critical process identification and control strategy development represent core competencies in risk-based quality management that enable organizations to focus resources on processes with greatest potential impact on product quality. These competencies require deep process understanding, risk assessment capabilities, and systematic approaches to control strategy optimization.
Continuous monitoring and trending analysis capabilities enable organizations to identify emerging quality risks before they impact product quality while providing data for systematic improvement of risk management effectiveness. These capabilities require data collection systems, analytical competencies, and management processes that translate monitoring results into proactive risk mitigation actions.
Supplier Management and Third-Party Risk Capabilities
Supplier management and third-party risk management represent critical organizational capabilities that directly impact product quality, regulatory compliance, and operational continuity. The complexity of pharmaceutical supply chains requires sophisticated approaches to supplier qualification, performance monitoring, and risk mitigation that go beyond traditional procurement practices.
Supplier qualification processes must assess not only technical capabilities but also quality culture, regulatory compliance history, and risk management effectiveness of potential suppliers. This assessment requires organizational capabilities in audit planning, execution, and reporting that provide confidence in supplier ability to meet pharmaceutical quality requirements consistently.
Performance monitoring systems must track supplier compliance with quality requirements, delivery performance, and responsiveness to quality issues over time. These systems require data collection capabilities, analytical competencies, and escalation processes that enable proactive management of supplier performance issues before they impact operations.
Risk mitigation strategies must address potential supply disruptions, quality failures, and regulatory compliance issues across the supplier network. Effective risk mitigation requires contingency planning, alternative supplier development, and inventory management strategies that maintain operational continuity while ensuring product quality.
The integration of supplier management with internal quality systems creates comprehensive quality assurance that extends across the entire value chain while maintaining accountability for product quality regardless of manufacturing location or supplier involvement. This integration requires organizational capabilities in supplier oversight, quality agreement management, and cross-functional coordination that ensure consistent quality standards throughout the supply network.
Implementation Roadmap for Cultural Risk Management Development
Staged Approach to Cultural Risk Management Development
The implementation of cultural risk management requires systematic, phased approach that builds organizational capabilities progressively while maintaining operational continuity and regulatory compliance. This staged approach recognizes that cultural transformation requires sustained effort over extended timeframes while providing measurable progress indicators that demonstrate value and maintain organizational commitment.
Phase 1: Foundation Building and Assessment establishes baseline understanding of current culture state, identifies immediate improvement opportunities, and creates infrastructure necessary for systematic cultural development. This phase includes comprehensive cultural assessment, leadership commitment establishment, initial training program development, and quick-win implementation that demonstrates early value from cultural investment.
Cultural assessment activities encompass employee surveys, management interviews, process observations, and regulatory compliance analysis that provide comprehensive understanding of current cultural strengths and improvement opportunities. These assessments establish baseline measurements that enable progress tracking while identifying specific areas requiring focused attention during subsequent phases.
Leadership commitment development ensures that senior management understands cultural transformation requirements, commits necessary resources, and demonstrates visible support for cultural change initiatives. This commitment includes resource allocation, communication of cultural expectations, and integration of cultural objectives into performance management systems.
Phase 2: Capability Development and System Implementation focuses on building specific competencies, implementing systematic processes, and creating organizational infrastructure that supports sustained cultural improvement. This phase includes comprehensive training program rollout, process improvement implementation, measurement system development, and initial culture champion network establishment.
Training program implementation provides employees with knowledge, skills, and tools necessary for effective participation in cultural transformation while creating shared understanding of quality expectations and risk management principles. These programs must be tailored to specific roles and responsibilities while maintaining consistency in core cultural messages.
Process improvement implementation creates systematic approaches to risk identification, assessment, and mitigation that embed cultural values into daily operations. These processes include structured problem-solving methodologies, escalation procedures, and continuous improvement practices that reinforce cultural expectations through routine operational activities.
Phase 3: Integration and Sustainment emphasizes cultural embedding, performance optimization, and continuous improvement capabilities that ensure long-term cultural effectiveness. This phase includes advanced measurement system implementation, culture champion network expansion, and systematic review processes that maintain cultural momentum over time.
Leadership Engagement Strategies for Sustainable Change
Leadership engagement represents the most critical factor in successful cultural transformation, requiring systematic strategies that ensure consistent leadership behavior, effective communication, and sustained commitment throughout the transformation process. Effective leadership engagement creates organizational conditions where cultural change becomes self-reinforcing while providing clear direction and resources necessary for transformation success.
Visible Leadership Commitment requires leaders to demonstrate cultural values through daily decisions, resource allocation priorities, and personal behavior that models expected cultural norms. This visibility includes regular communication of cultural expectations, participation in cultural activities, and recognition of employees who exemplify desired cultural behaviors.
Leadership communication strategies must provide clear, consistent messages about cultural expectations while demonstrating transparency in decision-making and responsiveness to employee concerns. Effective communication includes regular updates on cultural progress, honest discussion of challenges, and celebration of cultural achievements that reinforce the value of cultural investment.
Leadership Development Programs ensure that managers at all levels possess competencies necessary for effective cultural leadership including change management skills, coaching capabilities, and performance management approaches that support cultural transformation. These programs must be ongoing rather than one-time events to ensure sustained leadership effectiveness.
Change management competencies enable leaders to guide employees through cultural transformation while addressing resistance, maintaining morale, and sustaining momentum throughout extended change processes. These competencies include stakeholder engagement, communication planning, and resistance management approaches that facilitate smooth cultural transitions.
Accountability Systems ensure that leaders are held responsible for cultural outcomes within their areas of responsibility while providing support and resources necessary for cultural success. These systems include cultural metrics integration into performance management systems, regular cultural assessment processes, and recognition programs that reward effective cultural leadership.
Training and Development Frameworks
Comprehensive training and development frameworks provide employees with competencies necessary for effective participation in risk-based quality culture while creating organizational learning capabilities that support continuous cultural improvement. These frameworks must be systematic, role-specific, and continuously updated to reflect evolving regulatory requirements and organizational capabilities.
Foundational Training Programs establish basic understanding of quality principles, risk management concepts, and regulatory requirements that apply to all employees regardless of specific role or function. This training creates shared vocabulary and understanding that enables effective cross-functional collaboration while ensuring consistent application of cultural principles.
Quality fundamentals training covers basic concepts including customer focus, process thinking, data-driven decision making, and continuous improvement that form the foundation of quality culture. This training must be interactive, practical, and directly relevant to employee daily responsibilities to ensure engagement and retention.
Risk management training provides employees with capabilities in risk identification, assessment, communication, and escalation that enable proactive risk management throughout operations. This training includes both conceptual understanding and practical tools that employees can apply immediately in their work environment.
Role-Specific Advanced Training develops specialized competencies required for specific positions while maintaining alignment with overall cultural objectives and organizational quality strategy. This training addresses technical competencies, leadership skills, and specialized knowledge required for effective performance in specific roles.
Management training focuses on leadership competencies, change management skills, and performance management approaches that support cultural transformation while achieving operational objectives. This training must be ongoing and include both formal instruction and practical application opportunities.
Technical training ensures that employees possess current knowledge and skills required for effective job performance while maintaining awareness of evolving regulatory requirements and industry best practices. This training includes both initial competency development and ongoing skill maintenance programs.
Continuous Learning Systems create organizational capabilities for identifying training needs, developing training content, and measuring training effectiveness that ensure sustained competency development over time. These systems include needs assessment processes, content development capabilities, and effectiveness measurement approaches that continuously improve training quality.
Metrics and KPIs for Tracking Capability Maturation
Comprehensive measurement systems for cultural capability maturation provide objective evidence of progress while identifying areas requiring additional attention and investment. These measurement systems must balance quantitative indicators with qualitative assessments to capture the full scope of cultural development while providing actionable insights for continuous improvement.
Leading Indicators measure cultural inputs and activities that predict future cultural performance including training completion rates, employee engagement scores, participation in improvement activities, and leadership behavior assessments. These indicators provide early warning of cultural issues while demonstrating progress in cultural development activities.
Employee engagement measurements capture employee commitment to organizational objectives, satisfaction with work environment, and confidence in organizational leadership that directly influence cultural effectiveness. These measurements include regular survey processes, focus group discussions, and exit interview analysis that provide insights into employee perspectives on cultural development.
Training effectiveness indicators track not only completion rates but also competency development, knowledge retention, and application of training content in daily work activities. These indicators ensure that training investments translate into improved job performance and cultural behavior.
Lagging Indicators measure cultural outcomes including quality performance, regulatory compliance, operational efficiency, and customer satisfaction that reflect the ultimate impact of cultural investments. These indicators provide validation of cultural effectiveness while identifying areas where cultural development has not yet achieved desired outcomes.
Quality performance metrics include deviation rates, customer complaints, product recalls, and regulatory observations that directly reflect the effectiveness of quality culture in preventing quality issues. These metrics must be trended over time to identify improvement patterns and areas requiring additional attention.
Operational efficiency indicators encompass productivity measures, cost performance, delivery performance, and resource utilization that demonstrate the operational impact of cultural improvements. These indicators help demonstrate the business value of cultural investments while identifying opportunities for further improvement.
Integrated Measurement Systems combine leading and lagging indicators into comprehensive dashboards that provide management with complete visibility into cultural development progress while enabling data-driven decision making about cultural investments. These systems include automated data collection, trend analysis capabilities, and exception reporting that focus management attention on areas requiring intervention.
Benchmarking capabilities enable organizations to compare their cultural performance against industry standards and best practices while identifying opportunities for improvement. These capabilities require access to industry data, analytical competencies, and systematic comparison processes that inform cultural development strategies.
Future-Facing Implications for the Evolving Regulatory Landscape
Emerging Regulatory Trends and Capability Requirements
The regulatory landscape continues evolving toward increased emphasis on risk-based approaches, data integrity requirements, and organizational culture assessment that require corresponding evolution in organizational capabilities and management approaches. Organizations must anticipate these regulatory developments and proactively develop capabilities that address future requirements rather than merely responding to current regulations.
Enhanced Quality Culture Focus in regulatory inspections requires organizations to demonstrate not only technical compliance but also cultural effectiveness in sustaining quality performance over time. This trend requires development of cultural measurement capabilities, cultural audit processes, and systematic approaches to cultural development that provide evidence of cultural maturity to regulatory inspectors.
Risk-based inspection approaches focus regulatory attention on areas with greatest potential risk while requiring organizations to demonstrate effective risk management capabilities throughout their operations. This evolution requires mature risk assessment capabilities, comprehensive risk mitigation strategies, and systematic documentation of risk management effectiveness.
Technology Integration and Cultural Adaptation
Technology integration in pharmaceutical manufacturing creates new opportunities for operational excellence while requiring cultural adaptation that maintains human oversight and decision-making capabilities in increasingly automated environments. Organizations must develop cultural approaches that leverage technology capabilities while preserving the human judgment and oversight essential for quality decision-making.
Digital quality systems enable real-time monitoring, advanced analytics, and automated decision support that enhance quality management effectiveness while requiring new competencies in system operation, data interpretation, and technology-assisted decision making. Cultural adaptation must ensure that technology enhances rather than replaces human quality oversight capabilities.
Data Integrity in Digital Environments requires sophisticated understanding of electronic systems, data governance principles, and cybersecurity requirements that go beyond traditional paper-based quality systems. Cultural development must emphasize data integrity principles that apply across both electronic and paper systems while building competencies in digital data management.
Building Adaptive Organizational Capabilities
The increasing pace of change in regulatory requirements, technology capabilities, and market conditions requires organizational capabilities that enable rapid adaptation while maintaining operational stability and quality performance. These adaptive capabilities must be embedded in organizational culture and management systems to ensure sustained effectiveness across changing conditions.
Learning Organization Capabilities enable systematic capture, analysis, and dissemination of knowledge from operational experience, regulatory changes, and industry developments that inform continuous organizational improvement. These capabilities include knowledge management systems, learning processes, and cultural practices that promote organizational learning and adaptation.
Scenario planning and contingency management capabilities enable organizations to anticipate potential future conditions and develop response strategies that maintain operational effectiveness across varying circumstances. These capabilities require analytical competencies, strategic planning processes, and risk management approaches that address uncertainty systematically.
Change Management Excellence encompasses systematic approaches to organizational change that minimize disruption while maximizing adoption of new capabilities and practices. These capabilities include change planning, stakeholder engagement, communication strategies, and performance management approaches that facilitate smooth organizational transitions.
Resilience building requires organizational capabilities that enable sustained performance under stress, rapid recovery from disruptions, and systematic strengthening of organizational capabilities based on experience with challenges. These capabilities encompass redundancy planning, crisis management, business continuity, and systematic approaches to capability enhancement based on lessons learned.
The future pharmaceutical manufacturing environment will require organizations that combine operational excellence with adaptive capability, regulatory intelligence with proactive compliance, and technical competence with robust quality culture. Organizations successfully developing these integrated capabilities will achieve sustainable competitive advantage while contributing to improved patient outcomes through reliable access to high-quality pharmaceutical products.
The strategic integration of risk management practices with cultural transformation represents not merely an operational improvement opportunity but a fundamental requirement for sustained success in the evolving pharmaceutical manufacturing environment. Organizations implementing comprehensive risk buy-down strategies through systematic capability development will emerge as industry leaders capable of navigating regulatory complexity while delivering consistent value to patients, stakeholders, and society.
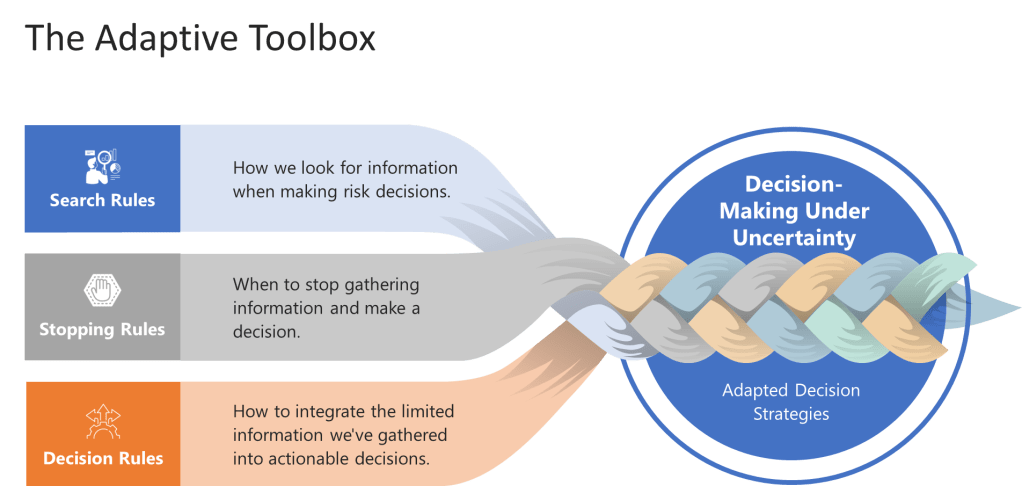
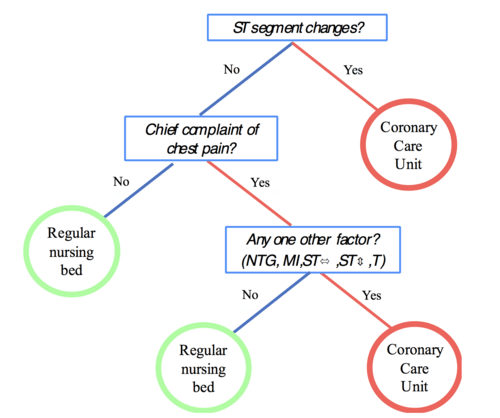
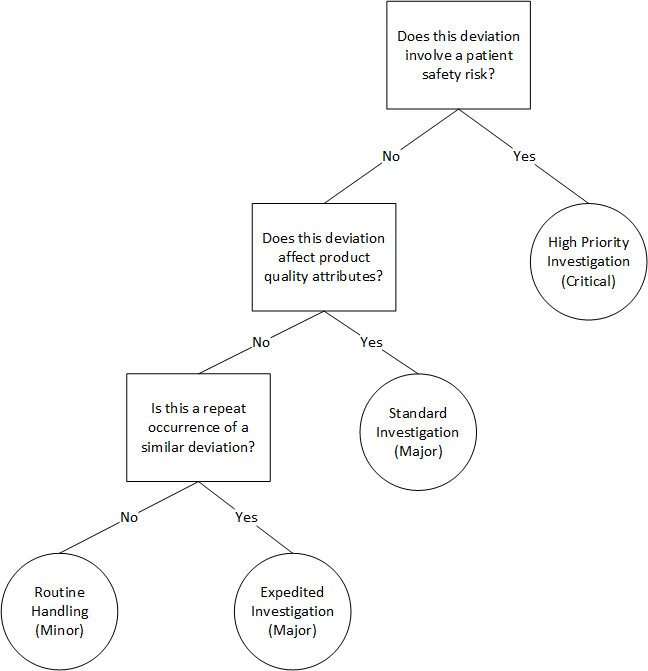
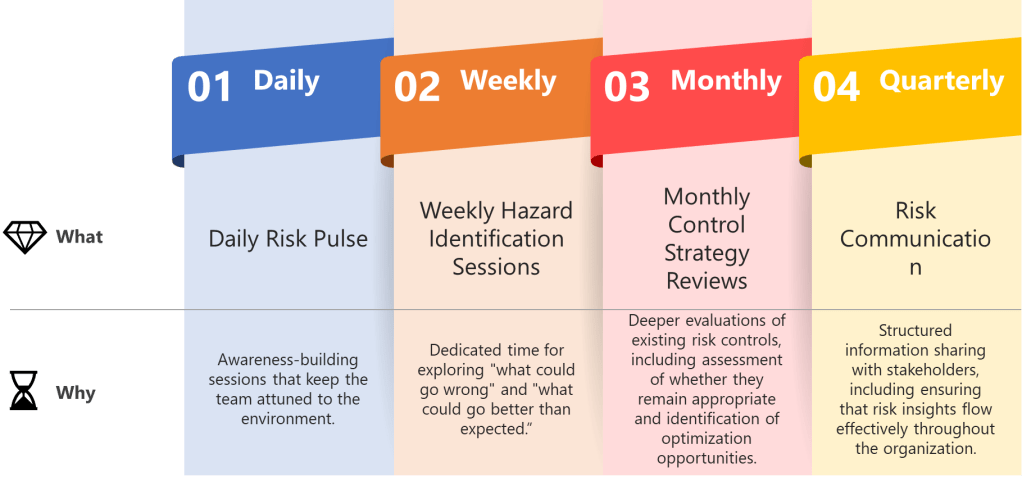
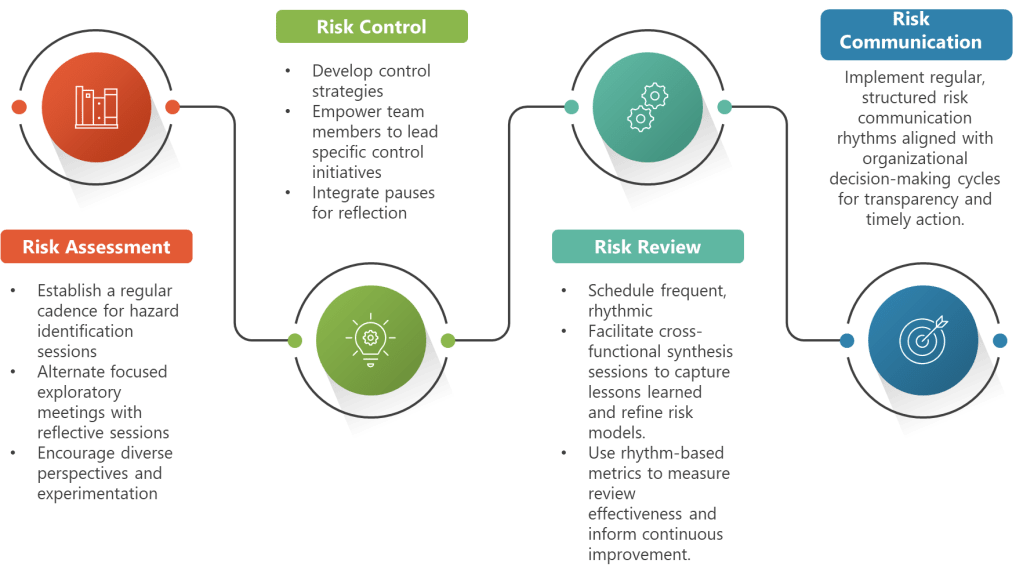

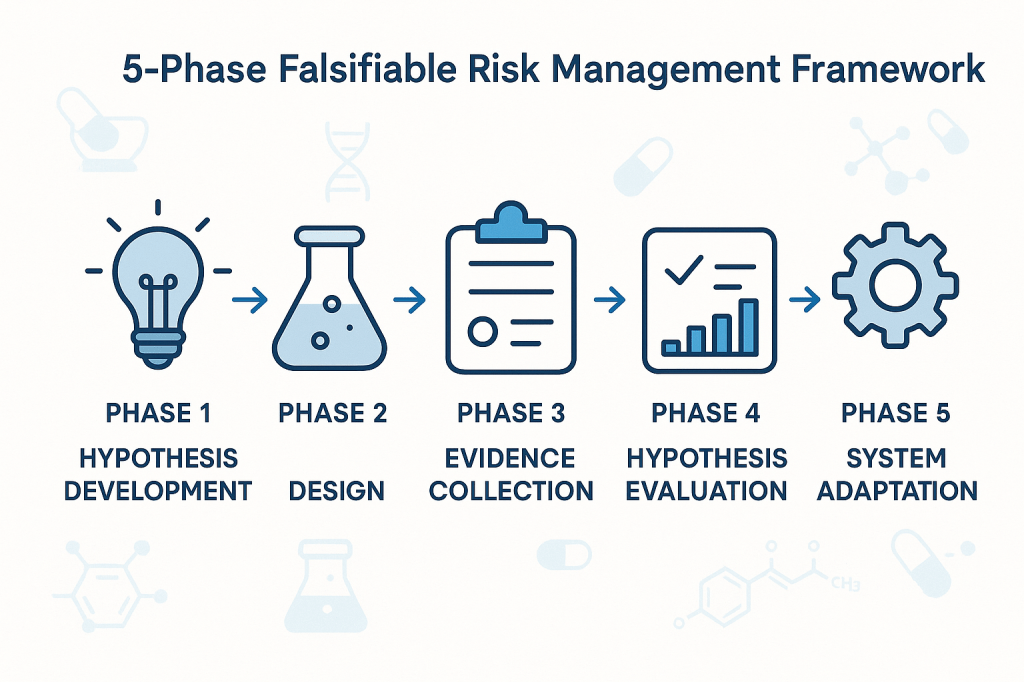
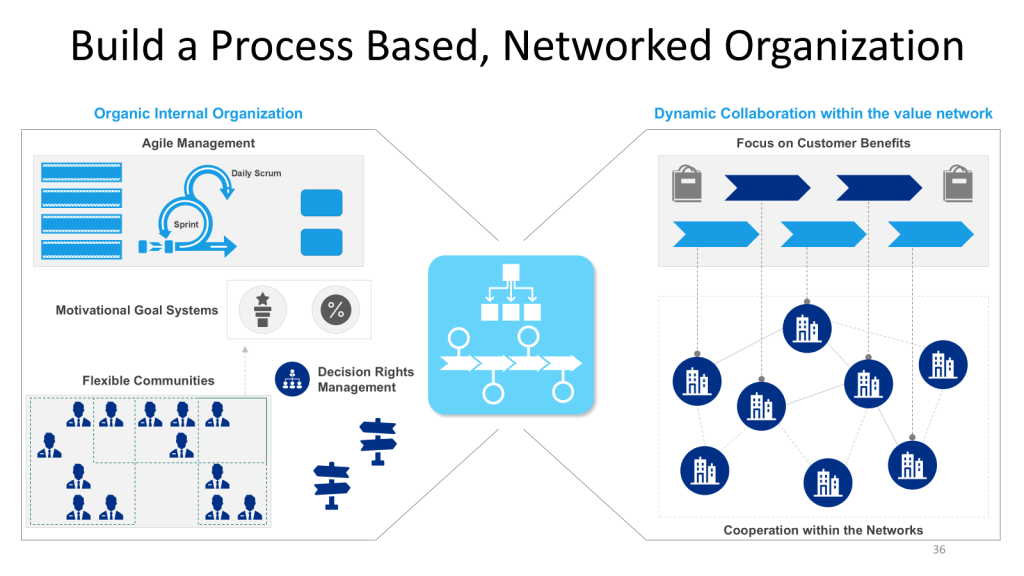
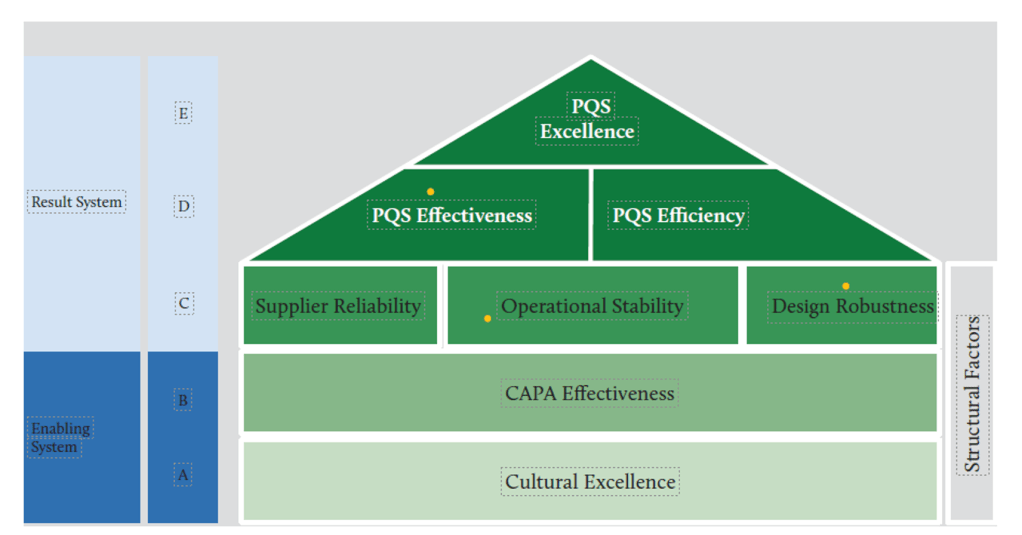
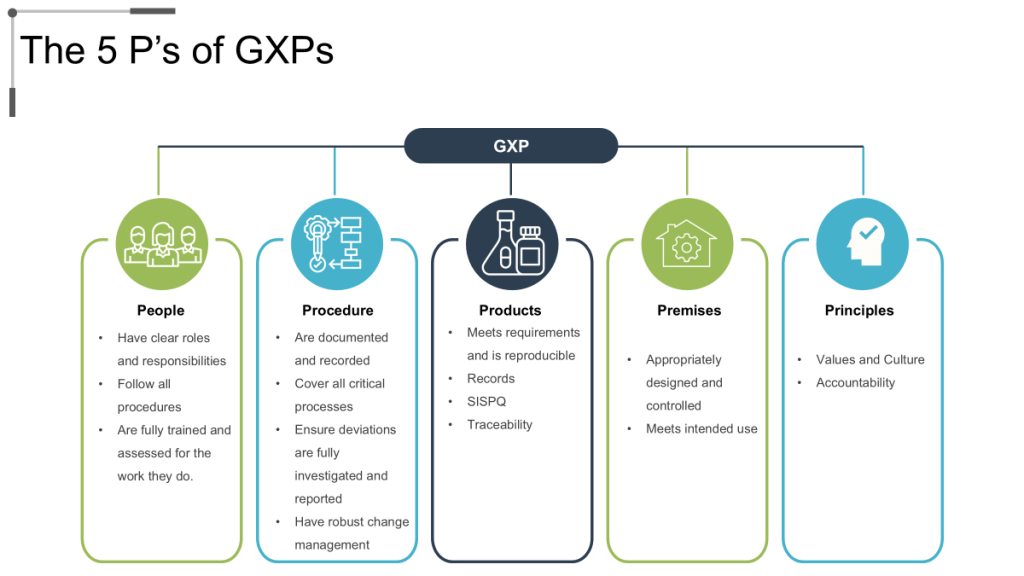
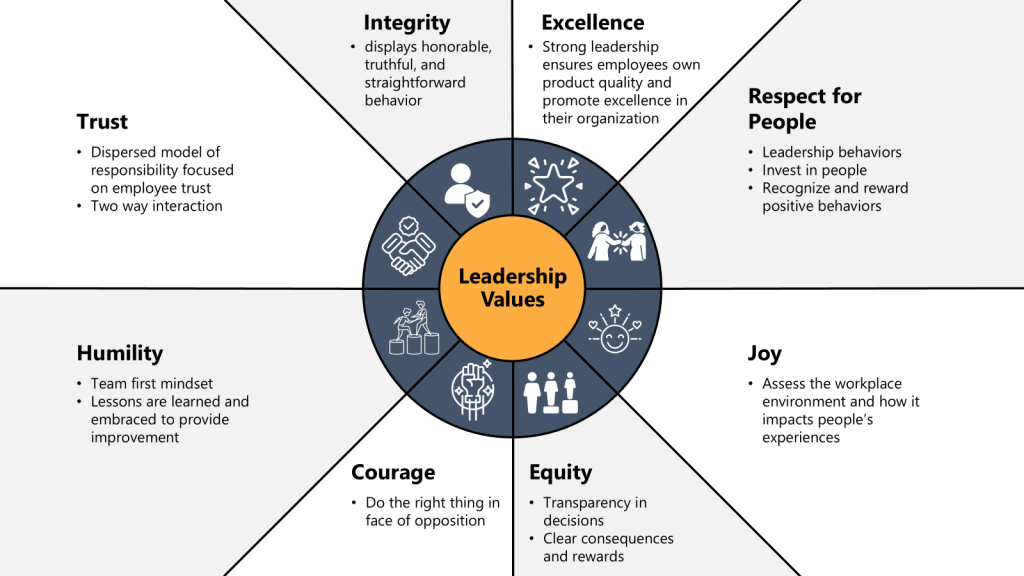

{kind=link}